
Pediatric Surgery — MCQs

On this page
Regarding ectopia vesicae, which of the following is true EXCEPT?
What is the surgical procedure for hypertrophic pyloric stenosis of infancy?
Hypochloremia, hypokalemia, and alkalosis are typically seen in which of the following conditions?
Which of the following causes distension of the abdomen?
What is the most likely cause of this physical finding?
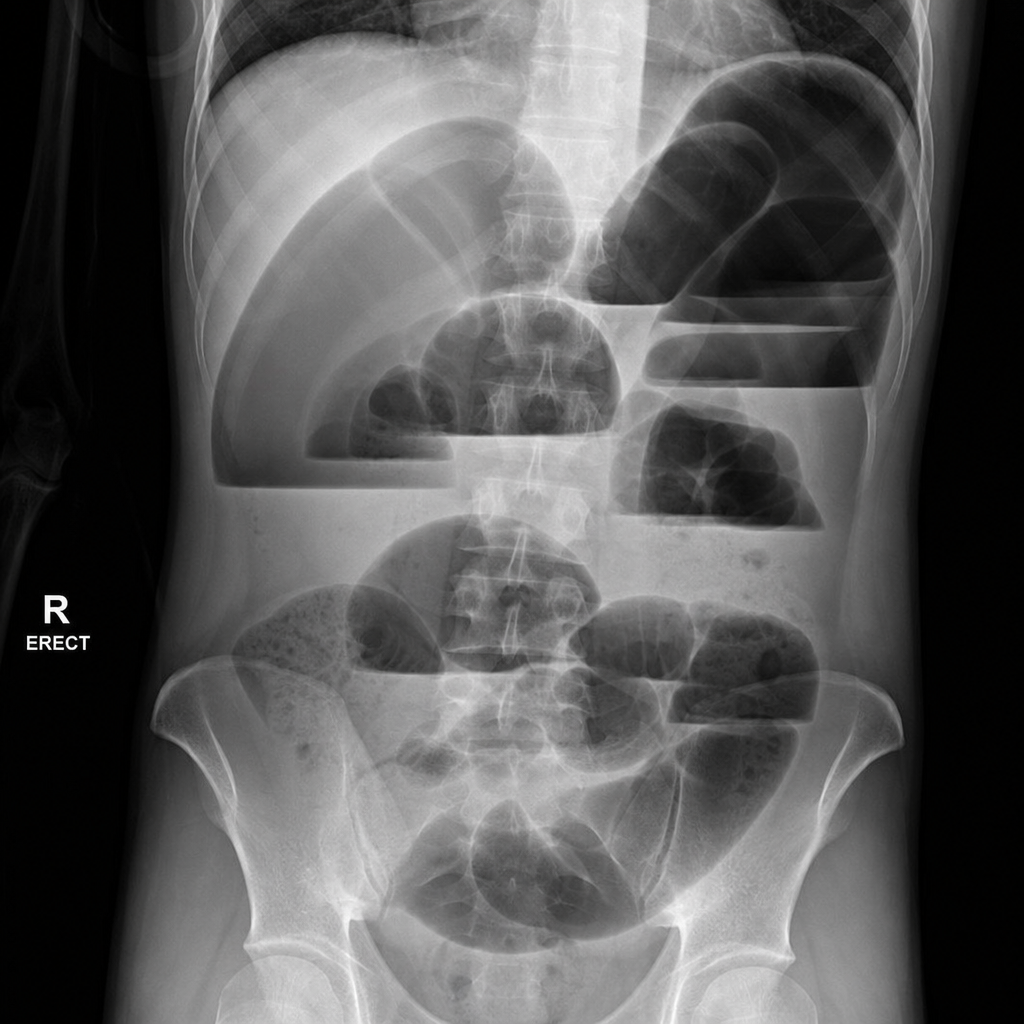
Regarding ectopia vesicae, which of the following statements is true except?
Which of the following statements about hypospadias is/are true?
True statements about congenital megacolon include all of the following except:
In cryptorchidism, what is the hallmark age for histological changes to appear in the testis?
What is the most common type of imperforate anus?
Practice by Chapter
Neonatal Physiology
Practice Questions
Congenital Anomalies Overview
Practice Questions
Neonatal Intestinal Obstruction
Practice Questions
Necrotizing Enterocolitis
Practice Questions
Hirschsprung's Disease
Practice Questions
Anorectal Malformations
Practice Questions
Pediatric Hernias
Practice Questions
Pyloric Stenosis
Practice Questions
Biliary Atresia
Practice Questions
Pediatric Tumors
Practice Questions
Congenital Diaphragmatic Hernia
Practice Questions
Pediatric Trauma
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app