
Renal Physiology — MCQs

On this page
What renal function is represented by the formula UV/P?
What is the typical Glomerular Filtration Rate (GFR) in a healthy adult?
Which of the following statements is true about GFR? GFR-Glomerular filtration rate RPF-Renal Plasma Flow
Which among the following is true regarding creatinine clearance?
Which of the following is not a mechanism of action of ADH?
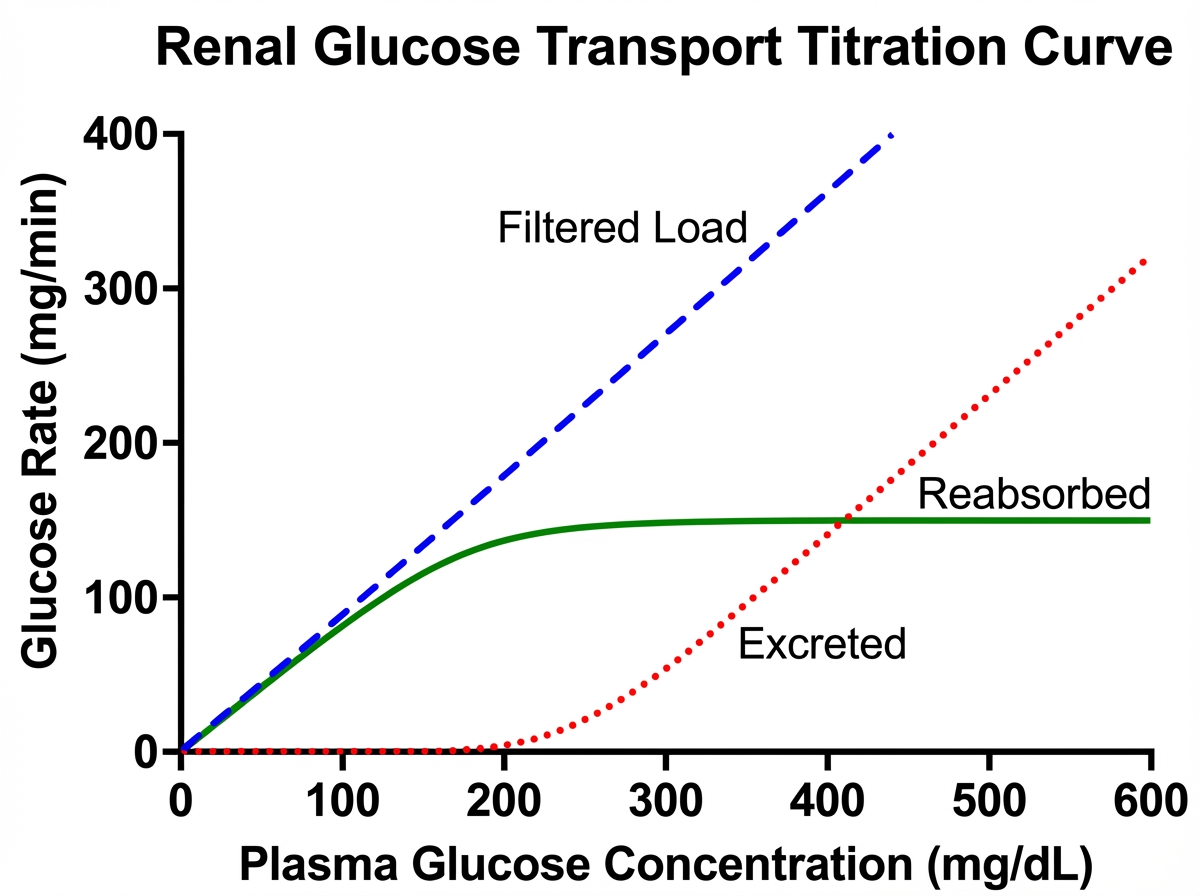
A patient has a blood glucose level of $200 \mathrm{mg} \%$ and GFR of 90. The transport maximum of the patient is as shown in the picture given below. What is the amount of glucose excreted?
Which of the following will occur on efferent arteriolar constriction?
All of the following substances have decreased concentration on the luminal side of the proximal convoluted tubule except:
Which region of the nephron reabsorbs the highest percentage of filtered bicarbonate?
Which mechanism primarily regulates sodium reabsorption in the collecting duct?
Practice by Chapter
Renal Blood Flow and Glomerular Filtration
Practice Questions
Tubular Reabsorption and Secretion
Practice Questions
Concentration and Dilution of Urine
Practice Questions
Acid-Base Regulation by the Kidneys
Practice Questions
Sodium and Water Balance
Practice Questions
Potassium Regulation
Practice Questions
Calcium and Phosphate Handling
Practice Questions
Micturition Physiology
Practice Questions
Renal Function Tests
Practice Questions
Integrative Responses to Fluid Challenges
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app