
Pharmacogenomics — MCQs

On this page
A 57-year-old man with myocardial infarction is admitted in cardiac emergency. Which of the following drugs might cause unexpected results based on the patient's CYP2C19 genotype?
Hemolysis in glucose-6-phosphate dehydrogenase enzyme deficiency may occur with all of the following drugs except:
Which drug does NOT cause hemolysis in G6PD deficiency?
Which drug does NOT cause hemolysis in a patient with G6PD deficiency?
Which of the following drugs can cause hemolysis in patients with Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency?
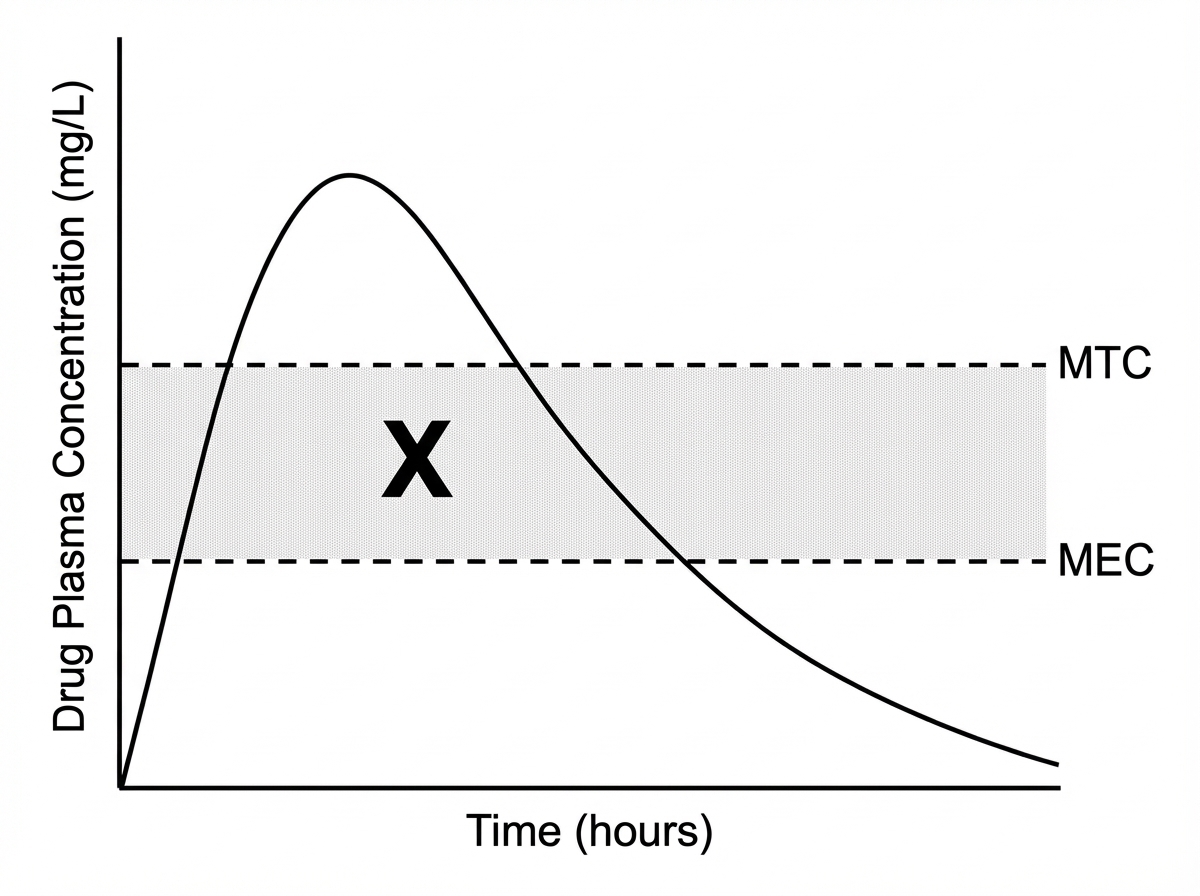
What does X represent in the given graph?
A patient with history of ischemic stroke was started on clopidogrel. However, she had another attack of stroke after 6 months. Which of the following is likely to be responsible for the failure of clopidogrel in this patient?
Prolonged apnea may occur in patients with a genetically determined abnormal variant of cholinesterase following intravenous administration of
Which enzyme deficiency is associated with severe reactions to sulfonamides in HIV patients?
Best predictor of varenicline response in smoking cessation is:
Practice by Chapter
Genetic Basis of Drug Response
Practice Questions
Pharmacogenomic Testing
Practice Questions
Cytochrome P450 Polymorphisms
Practice Questions
Pharmacogenomics of Drug Transporters
Practice Questions
Pharmacogenomics in Oncology
Practice Questions
Pharmacogenomics in Cardiovascular Therapeutics
Practice Questions
Pharmacogenomics in Psychiatry
Practice Questions
Personalized Medicine Approaches
Practice Questions
Ethical Considerations in Pharmacogenomics
Practice Questions
Implementation of Pharmacogenomics in Clinical Practice
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app