
General Pharmacology — MCQs

On this page
Which of the following is FALSE regarding the sublingual route of drug administration?
Which of the following is NOT a prodrug?
Which of the following is a prodrug?
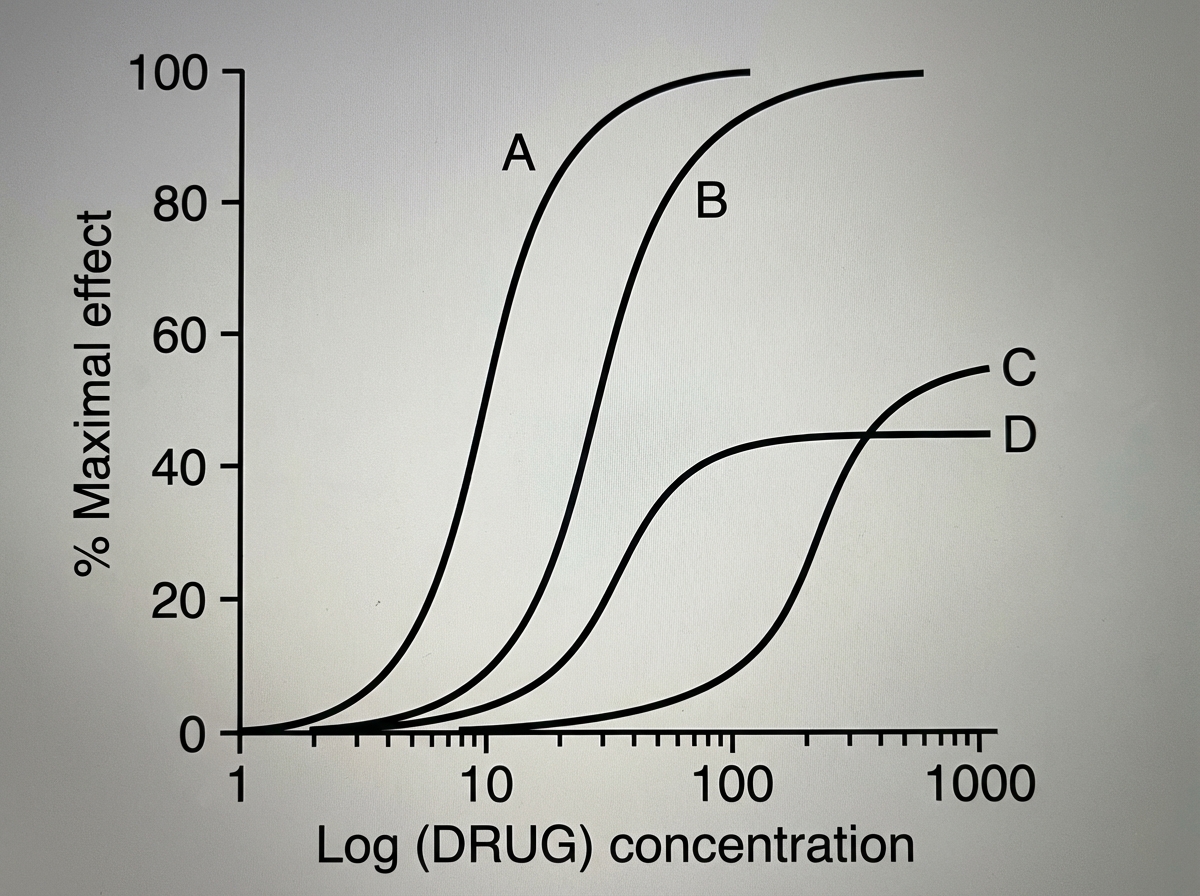
True regarding the following is?
Which of the following is a CYP-450 inducer?
Phase 4 clinical trials are carried out:
What advice should be given to a lactating mother regarding drug intake?
Which of the following drugs does NOT cause enzyme inhibition?
In new drug designing, what problem arises?
All of the following drugs act on ionic channels except?
Practice by Chapter
Pharmacokinetics: Absorption and Distribution
Practice Questions
Pharmacokinetics: Metabolism and Excretion
Practice Questions
Pharmacodynamics and Receptor Theory
Practice Questions
Drug-Receptor Interactions and Dose-Response
Practice Questions
Pharmacogenetics and Personalized Medicine
Practice Questions
Adverse Drug Reactions and Toxicity
Practice Questions
Drug Interactions
Practice Questions
Drug Development and Regulation
Practice Questions
Pediatric and Geriatric Pharmacology
Practice Questions
Placental Transfer and Lactation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app