
General Pharmacology — MCQs

On this page
Which of the following is FALSE regarding the sublingual route of drug administration?
Which of the following is NOT a prodrug?
Which of the following is a prodrug?
What advice should be given to a lactating mother regarding drug intake?
What is true about inverse agonism?
Which of the following drugs is an enzyme inducer?
Which of the following is an alkaloid?
According to the Drugs and Cosmetics Rules, match the following medications with their corresponding schedules: 1. Insulin, 2. Hepatitis B vaccine, 3. Morphine, 4. Veterinary drugs. Schedules: A. Schedule H, B. Schedule Z, C. Schedule G, D. Schedule X.
Which is correct about the marked area in the blister strip of Paracetamol (PCM)? (Recent NEET Pattern 2016-17)
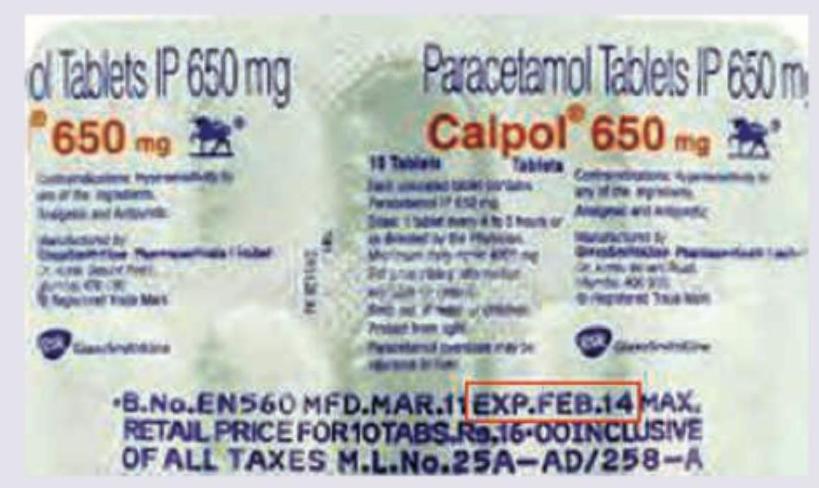
The image given below shows:

Practice by Chapter
Pharmacokinetics: Absorption and Distribution
Practice Questions
Pharmacokinetics: Metabolism and Excretion
Practice Questions
Pharmacodynamics and Receptor Theory
Practice Questions
Drug-Receptor Interactions and Dose-Response
Practice Questions
Pharmacogenetics and Personalized Medicine
Practice Questions
Adverse Drug Reactions and Toxicity
Practice Questions
Drug Interactions
Practice Questions
Drug Development and Regulation
Practice Questions
Pediatric and Geriatric Pharmacology
Practice Questions
Placental Transfer and Lactation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app