
Drug-Receptor Interactions and Dose-Response — MCQs

Which of the following is a G protein coupled receptor?
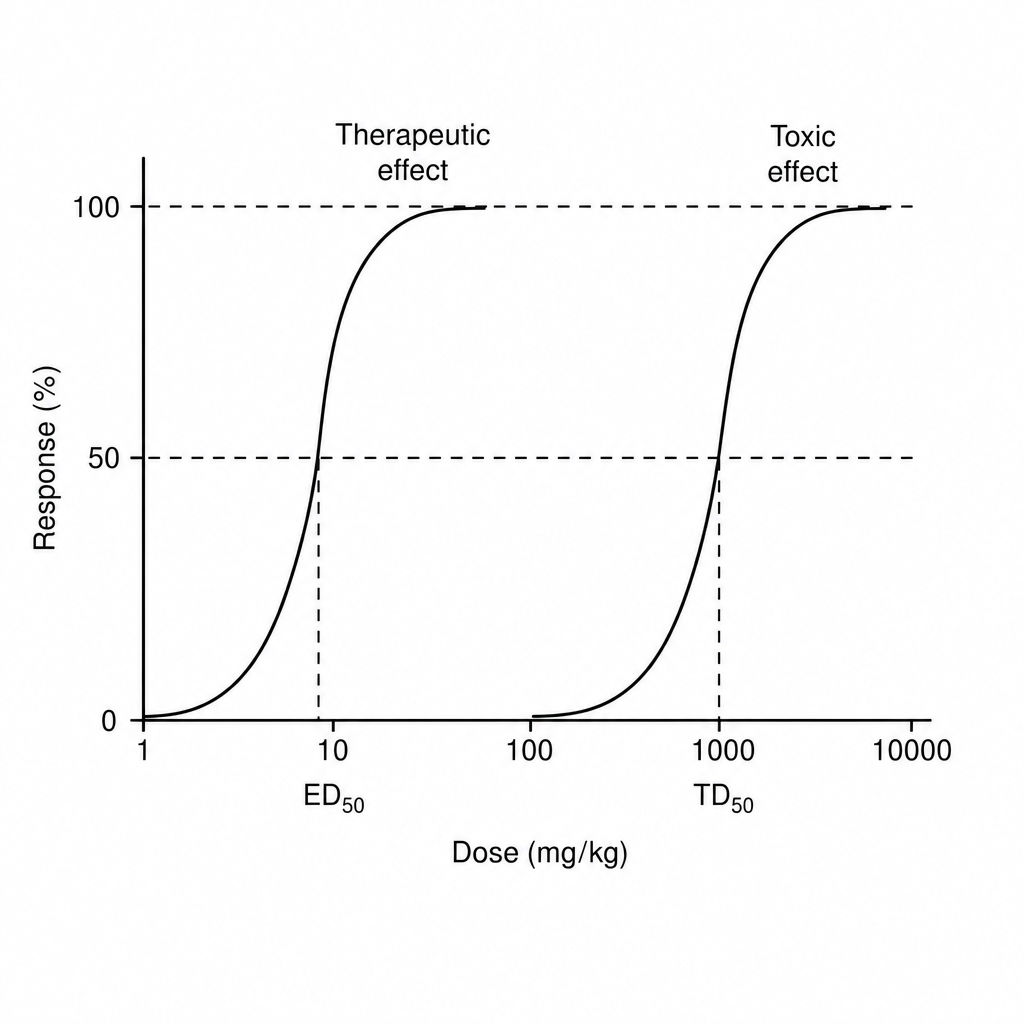
The therapeutic index of a drug is defined as the ratio between the toxic dose and the effective dose.
Variation in sensitivity of response to different doses of a drug in different individuals is obtained from?
Which of the following best demonstrates the variability in drug responsiveness among individuals?
With which of the following receptors does theophylline have an antagonistic interaction?
Find therapeutic index of drug from the information given below in the graph
Which one of the following is true for competitive antagonism?
Which of the following is false about drugs given in urinary incontinence
When two different chemicals act on two different receptors and their responses are opposite to each other on the same cell, this phenomenon is called?
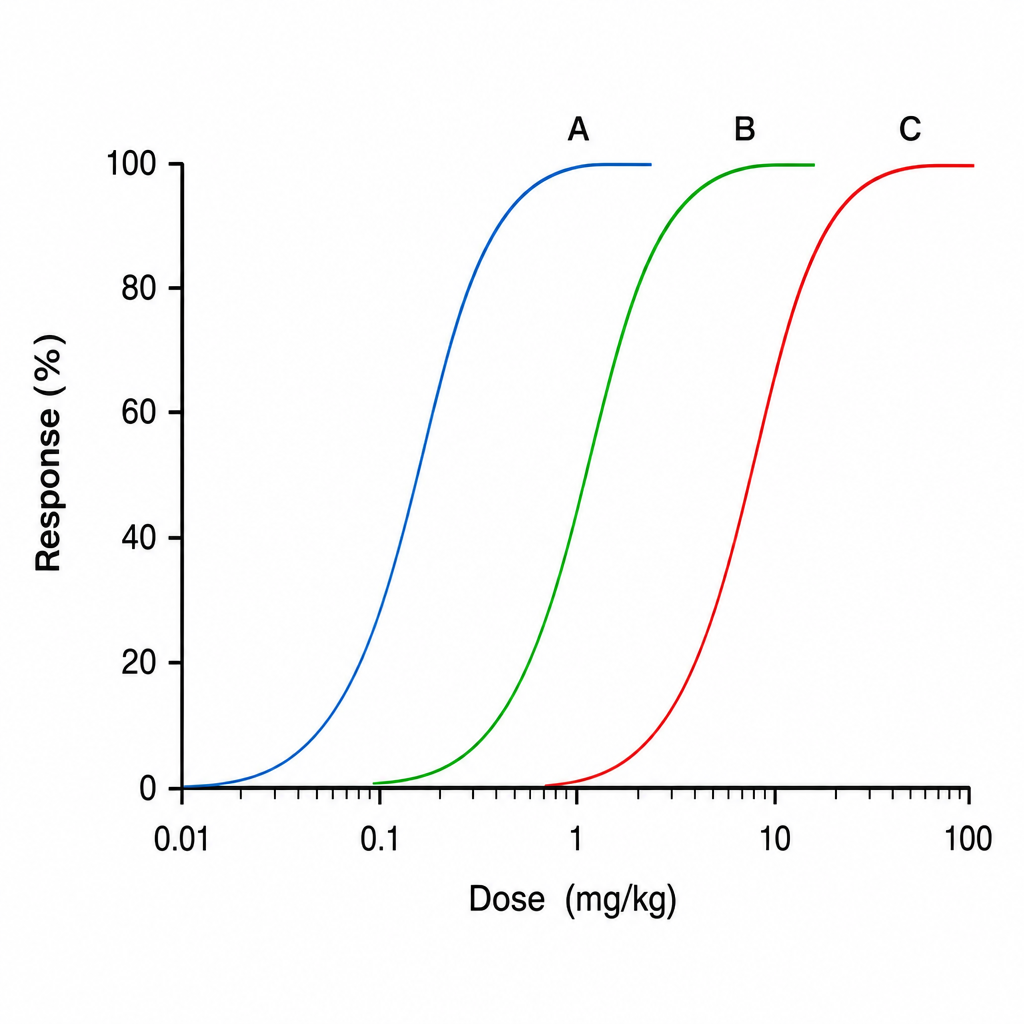
The graph below shows dose-response curves for three drugs A, B, and C. Which of the following drugs has the highest potency?
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app