
General Pharmacology — MCQs

On this page
All the following are ligand-gated ion channels EXCEPT:
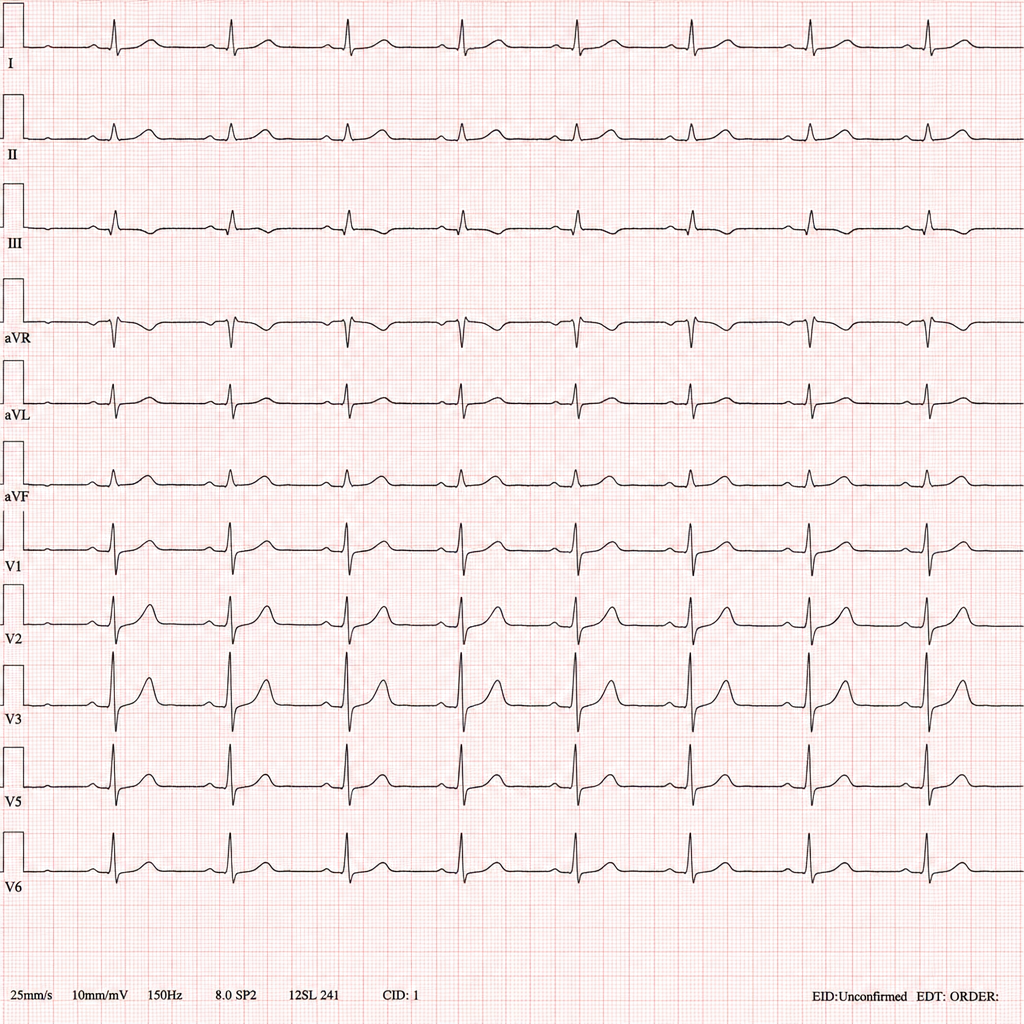
A 34-year-old male with opioid dependence enrolled in a maintenance programme presents to the emergency department with palpitations and two witnessed syncopal episodes over the past 24 hours. His only medication was started 6 weeks ago at a community addiction clinic. Baseline ECG on enrolment was normal. The on-call resident records the ECG shown in Image 1. Which drug is most likely responsible for the rhythm disturbance seen?
Which of the following drugs can be administered through all routes?
Therapeutic drug monitoring is done for all of the following drugs except?
Which of the following is an example of physiological antagonism?
Which drug schedule requires medical supervision for its administration?
Phase I clinical trials are primarily conducted to assess what aspect of a new drug?
Type A (augmented) adverse drug reactions are characterized by all EXCEPT:
In which phase of clinical trials is the efficacy of a new drug compared with an existing standard drug?
Adrenaline, noradrenaline, and dopamine act through which type of receptor?
Practice by Chapter
Pharmacokinetics: Absorption and Distribution
Practice Questions
Pharmacokinetics: Metabolism and Excretion
Practice Questions
Pharmacodynamics and Receptor Theory
Practice Questions
Drug-Receptor Interactions and Dose-Response
Practice Questions
Pharmacogenetics and Personalized Medicine
Practice Questions
Adverse Drug Reactions and Toxicity
Practice Questions
Drug Interactions
Practice Questions
Drug Development and Regulation
Practice Questions
Pediatric and Geriatric Pharmacology
Practice Questions
Placental Transfer and Lactation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app