
Cardiovascular Drugs — MCQs

On this page
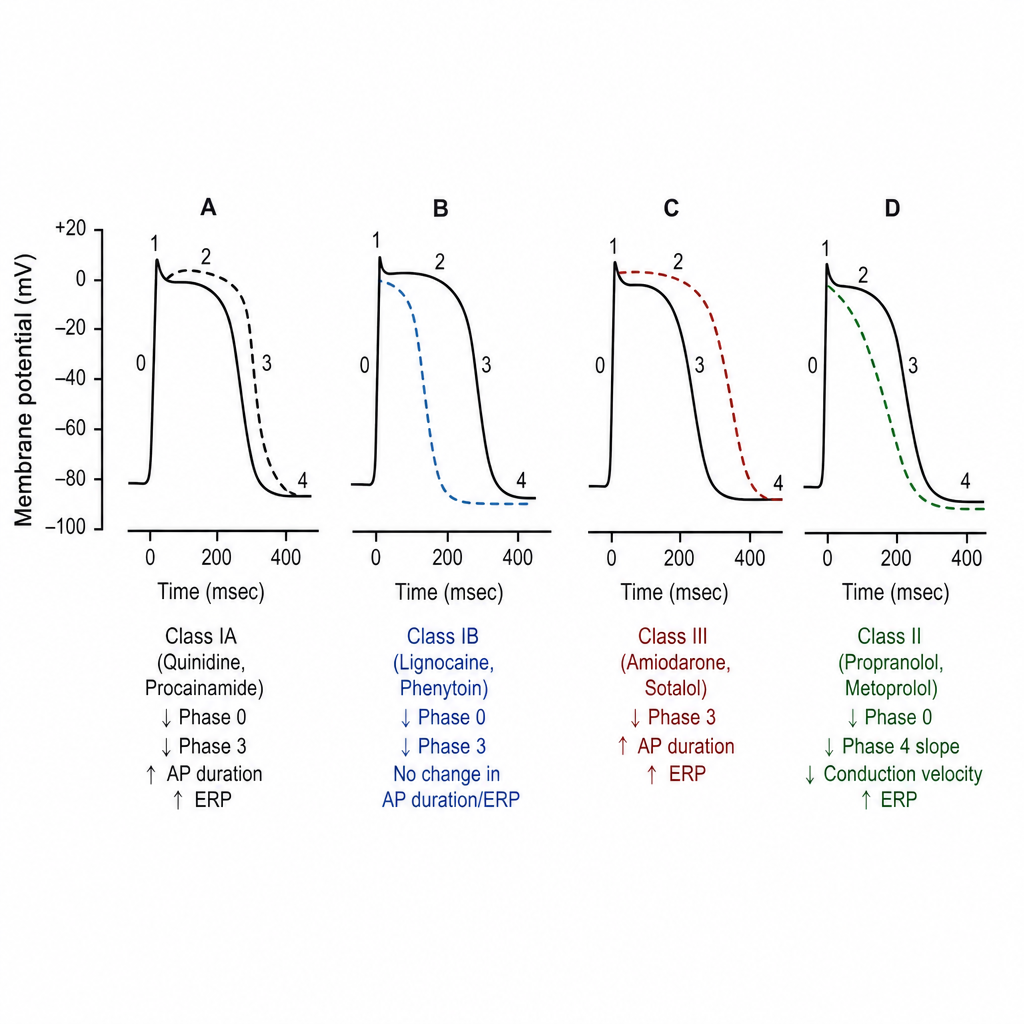
Which of the following graphs of cardiac action potential represents mechanism of action of lignocaine?
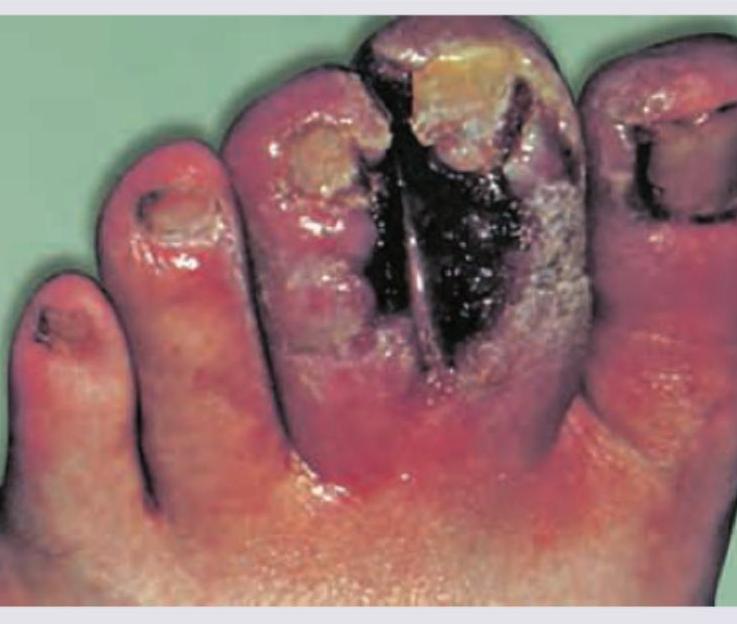
A 30-year-old male chronic smoker presents with pain in his legs on walking that is relieved by rest (stable intermittent claudication). Exercise therapy and smoking cessation have been initiated, but he continues to have limiting symptoms. Which of the following drugs is the preferred drug for improving walking distance in stable intermittent claudication according to major guidelines (e.g., ACC/AHA)?
A patient with rheumatic heart disease is in atrial fibrillation. The ECG shows irregularly irregular heart rhythm with absent P waves and down-sloping ST segment depression pronounced in V2-V3 and in the rhythm strip. The patient is experiencing general unwell being. Given the ECG findings and history, which medication is most likely causing the patient's symptoms?
Which one of the following responses to intravenous adenosine is correctly matched?
Which of the following anti-hypertensive drugs is/are best avoided during pregnancy ?
Match the following antiarrhythmic drugs with their mechanism of action: | Mechanism of action | Drug | | :-- | :-- | | 1. Na+ channel blocker | A. Quinidine | | 2. K+ channel blocker | B. Digoxin | | 3. Na+K+ ATPase inhibitor | C. Esmolol | | 4. Beta-blocker | D. Ibutilide |
Identify the correct match, regarding the drug and its adverse effect.
A patient who is on treatment for hyperlipidemia develops gallstones. What is the mechanism of action of the causative drug that was given to this patient?
Which among the following is the false statement regarding statins?
Preferred drug for the treatment of ventricular tachycardia is
Practice by Chapter
Antihypertensive Agents
Practice Questions
Drugs for Heart Failure
Practice Questions
Antiarrhythmic Drugs
Practice Questions
Antianginal Agents
Practice Questions
Lipid-Lowering Drugs
Practice Questions
Anticoagulants and Antiplatelet Drugs
Practice Questions
Thrombolytic Agents
Practice Questions
Drugs Used in Pulmonary Hypertension
Practice Questions
Drugs Used in Shock
Practice Questions
Cardiovascular Effects of Non-Cardiovascular Drugs
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app