
Oncology — MCQs

On this page
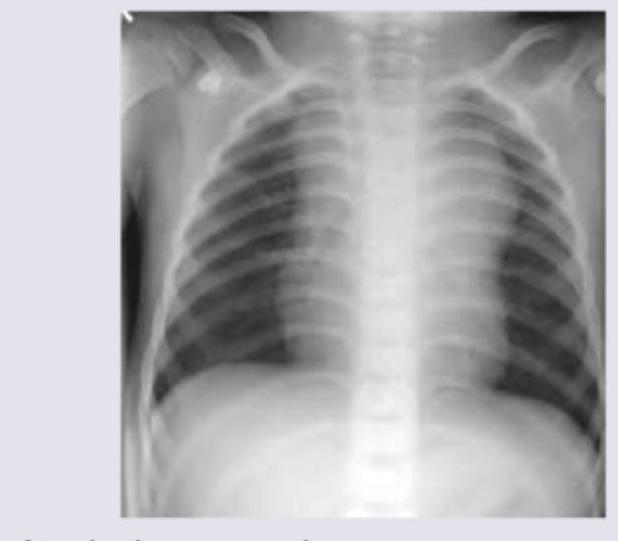
A 2-year-old boy presents with cough and breathlessness for last one month. On examination pallor is present and enlarged liver is palpable 3 cm below costal margin. CXR shows?
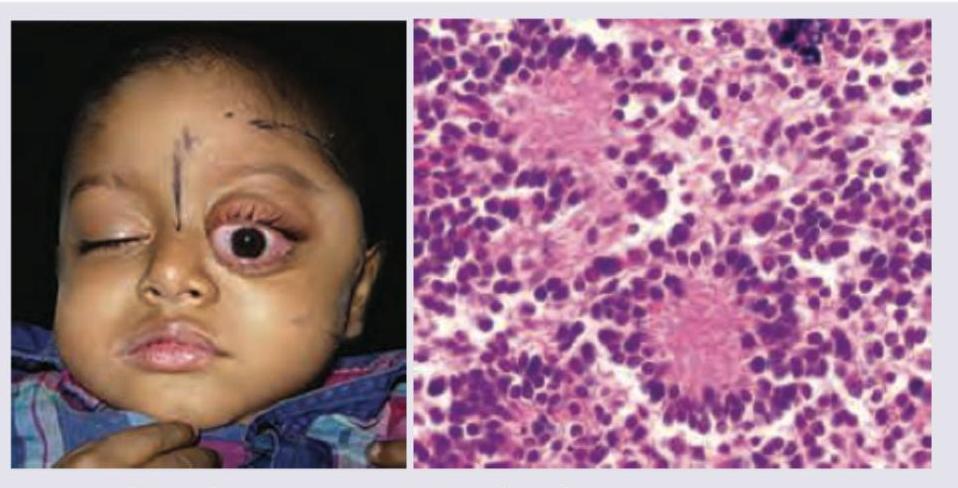
A 4-year-old child with rapidly growing swelling of left eye with loss of vision over last 4 weeks. On examination hepatosplenomegaly is noted. CBC shows anemia, hyperleukocytosis with low platelets. Bone marrow aspiration was performed. Diagnosis is?
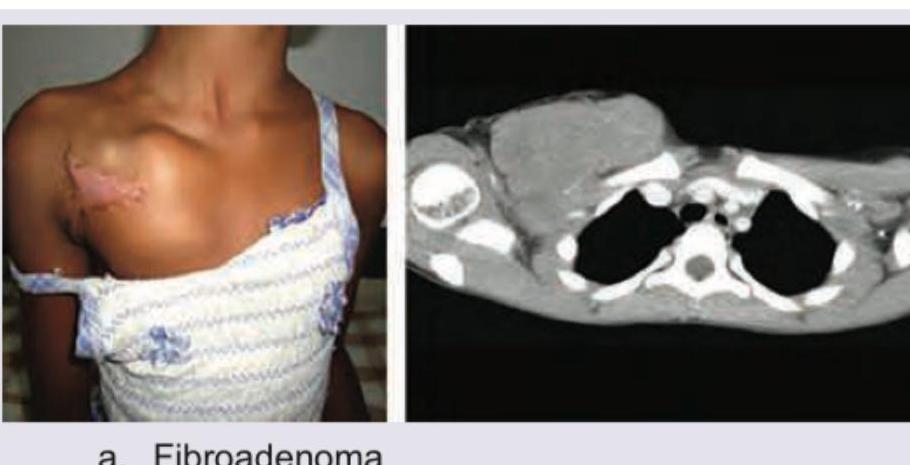
An 8-year-old girl presents with swelling over right anterior chest wall. Which is the correct diagnosis?
One-year-old child presents with abdominal lump. All are true about the condition shown below except:
A 6-month-old infant with a progressively increasing abdominal lump. Investigations revealed a stage IVs neuroblastoma. All are correct about the condition except:
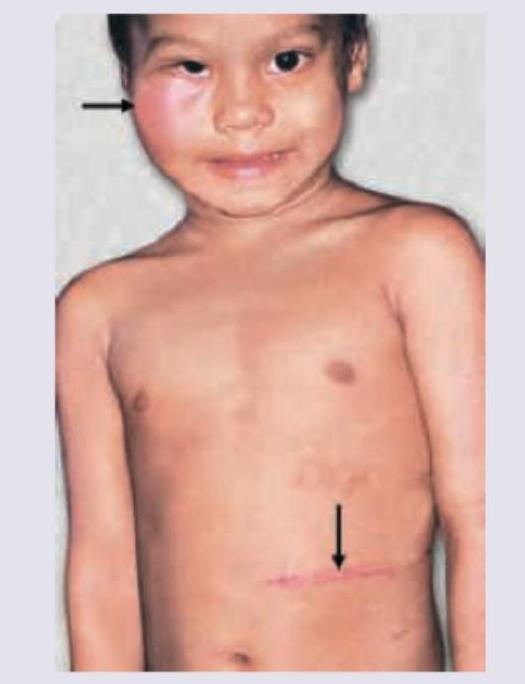
Comment on the diagnosis of this child, presenting with eye swelling. (Recent NEET Pattern 2016-17)
A mother notices a swelling in the abdomen of her 3-year-old child while bathing him. He had a history of hematuria two weeks back, which resolved spontaneously. On examination, a right-sided reniform ballotable mass was found. What is the most appropriate initial investigative approach?
Which of the following malignant diseases of children has the best prognosis -
Abdominal Ultrasonography in a 3 year old boy shows a solid circumscribed hypoechoic renal mass. Most likely diagnosis is-
Most common tumor in the part of the brain shown (arrow) among children is
Practice by Chapter
Leukemias
Practice Questions
Lymphomas
Practice Questions
CNS Tumors
Practice Questions
Neuroblastoma
Practice Questions
Wilms Tumor
Practice Questions
Rhabdomyosarcoma
Practice Questions
Bone Tumors
Practice Questions
Retinoblastoma
Practice Questions
Histiocytosis Syndromes
Practice Questions
Principles of Pediatric Chemotherapy
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Late Effects of Cancer Treatment
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app