
Oncology — MCQs

On this page
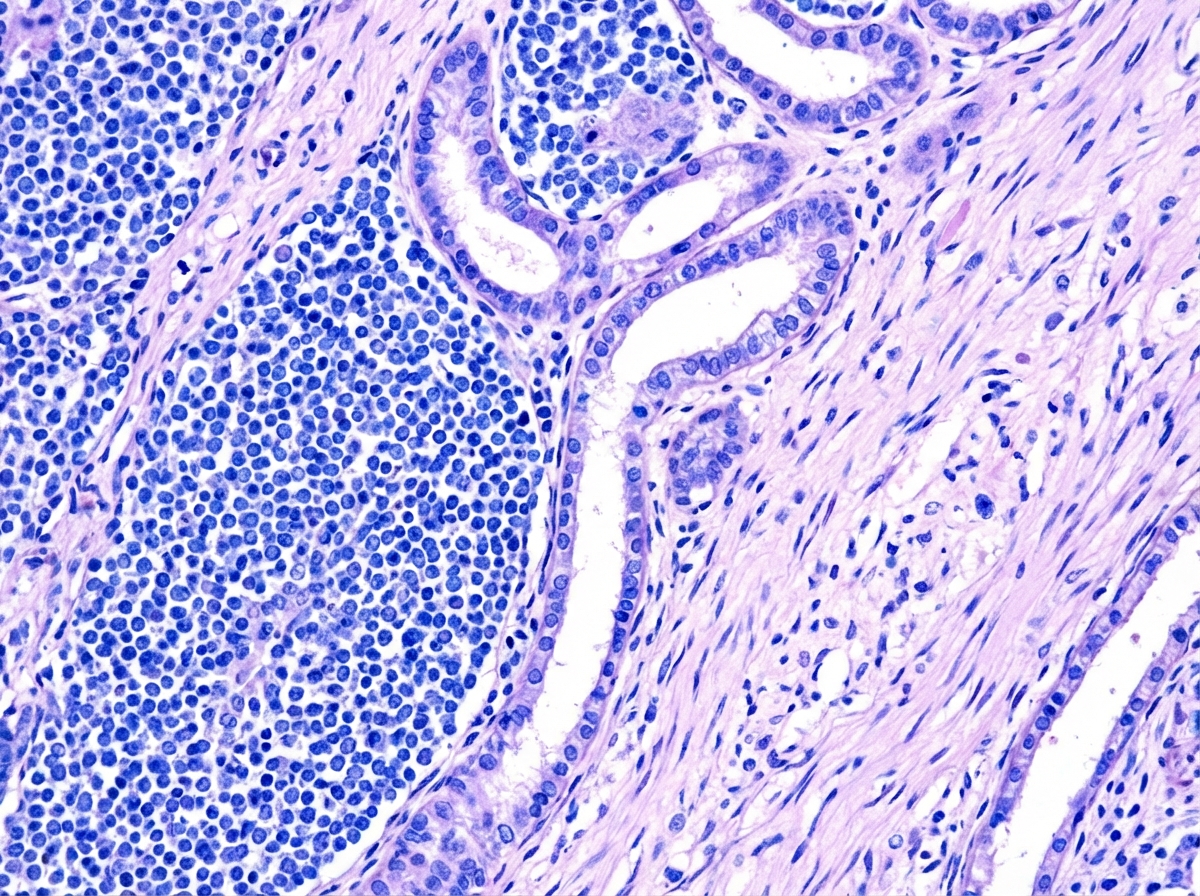
A 4-year-old child presented with a palpable, painless, and slowly increasing abdominal mass in the right flank region, accompanied by episodes of fever and hematuria. Examination revealed hypertension. Following a CT scan of the abdomen, the patient underwent surgical resection of the mass. The gross specimen and histopathological examination are provided. Which of the following drugs is NOT approved for the management of this condition?
WAGR syndrome includes all except?
What is the most common malignancy of the liver in children?
Wilms' tumour is associated with all of the following except?
Which of the following is the most important prognostic factor in ALL?
Practice by Chapter
Leukemias
Practice Questions
Lymphomas
Practice Questions
CNS Tumors
Practice Questions
Neuroblastoma
Practice Questions
Wilms Tumor
Practice Questions
Rhabdomyosarcoma
Practice Questions
Bone Tumors
Practice Questions
Retinoblastoma
Practice Questions
Histiocytosis Syndromes
Practice Questions
Principles of Pediatric Chemotherapy
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Late Effects of Cancer Treatment
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app