
Nephrology — MCQs

On this page
Following urine microscopy and examination, a child is found to have hematuria and RBC casts in the urine. What is the probable cause for this?
A 7-year-old child presents with generalized edema, frothy urine, and fatigue for 2 weeks. Laboratory findings show serum albumin 1.8 g/dL, total cholesterol 350 mg/dL, and 24-hour urine protein 5 g/day. Renal function is normal. Kidney biopsy shows normal glomeruli on light microscopy but effacement of podocyte foot processes on electron microscopy. What is the most appropriate initial treatment?
A 5-year-old child with chronic kidney disease (CKD) presents with bow legs. Laboratory investigations reveal: Serum Calcium: 9.1 mg/dL (normal) Serum Phosphate: 6.9 mg/dL (elevated) Alkaline Phosphatase: Elevated 25(OH) Vitamin D: Low What is the most appropriate next step in management?
A 12-year-old child is diagnosed with systemic lupus erythematosus (SLE) and presents with nephrotic-range proteinuria. Renal biopsy reveals "wire loop lesions." Which of the following is the drug of choice in this case?
A 1 month old child with recurrent episodes of fever. On examination a suprapubic swelling was noticed and mother reports poor urinary stream. MCUG was performed. All are true about the condition shown except:
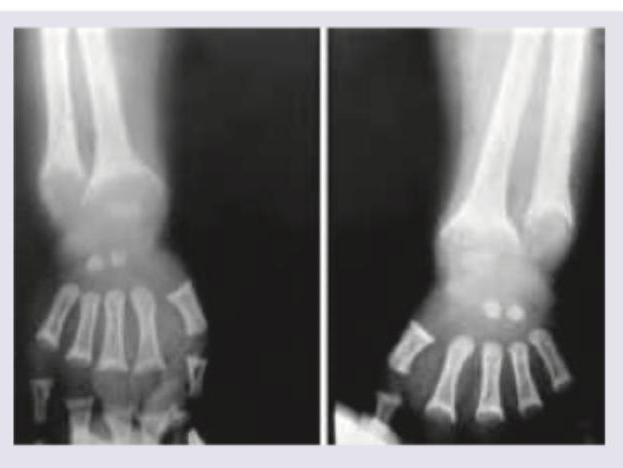
A 5-year-old boy from upper socioeconomic background presents with bowing of legs. On work up serum calcium was $9 \mathrm{mg} \%$ with serum phosphate of $1 \mathrm{mg} \%$ with normal serum alkaline phosphatase (ALP) and normal serum PTH. X-ray wrist joint is given. Comment on the diagnosis:
A child during evaluation of recurrent hematuria has the following eye finding. He has sensorineural deafness and history of similar illness in family members. What is the diagnosis?

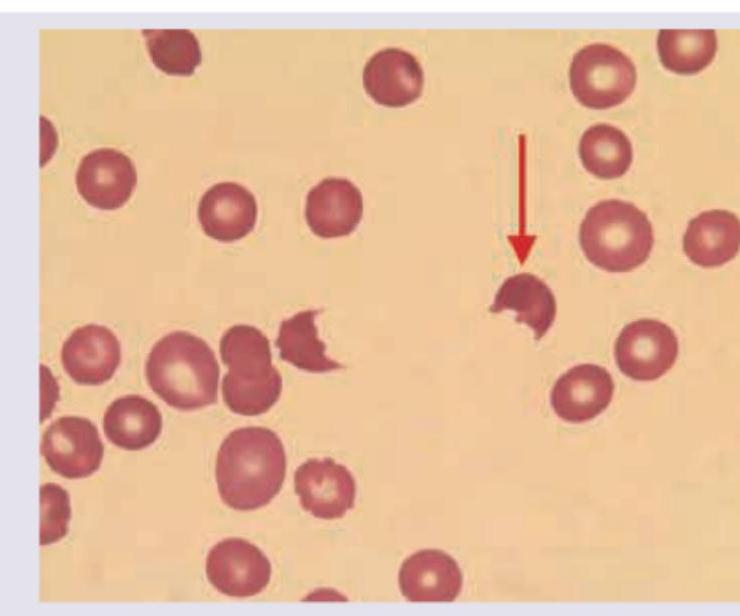
A child with history of diarrhea 1 week back presents with sudden onset pallor and oliguria. Peripheral smear findings are shown below. Which of the following is unlikely finding in this disorder?
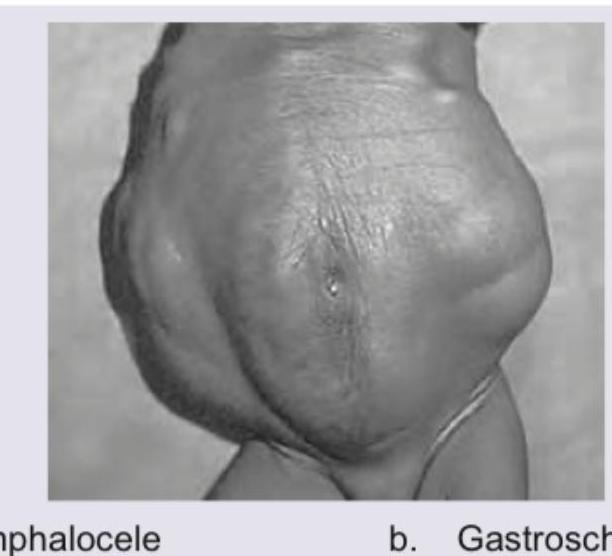
Identify this condition?
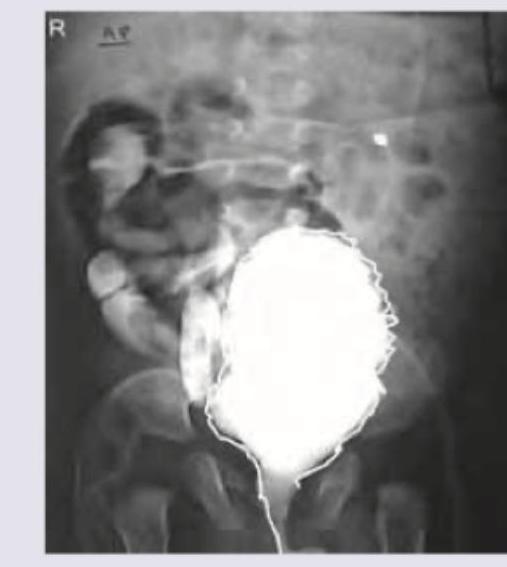
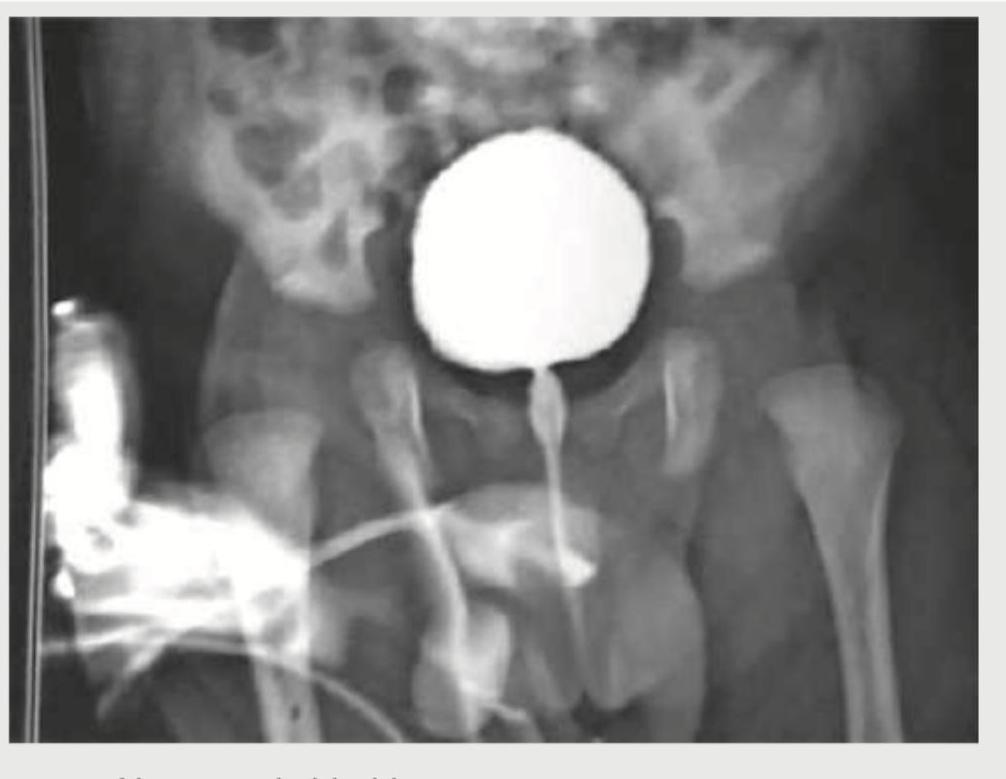
A 3-year-old boy presents with occasional episodes of passing urine after very long durations, going up to as long as 12 hours. Since the child cries due to discomfort and then passes urine with straining, a VCUG is ordered. The VCUG image is shown below. What is the diagnosis?
Practice by Chapter
Urinary Tract Infections
Practice Questions
Vesicoureteral Reflux
Practice Questions
Glomerulonephritis
Practice Questions
Nephrotic Syndrome
Practice Questions
Acute Kidney Injury
Practice Questions
Chronic Kidney Disease
Practice Questions
Renal Tubular Disorders
Practice Questions
Congenital Anomalies of the Kidney
Practice Questions
Hydronephrosis
Practice Questions
Hypertension in Children
Practice Questions
Hemolytic Uremic Syndrome
Practice Questions
Renal Replacement Therapy in Children
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app