
Immunology and Allergies — MCQs

On this page
In which of the following disorders, vaccines are not contraindicated in the person suffering from that disease?
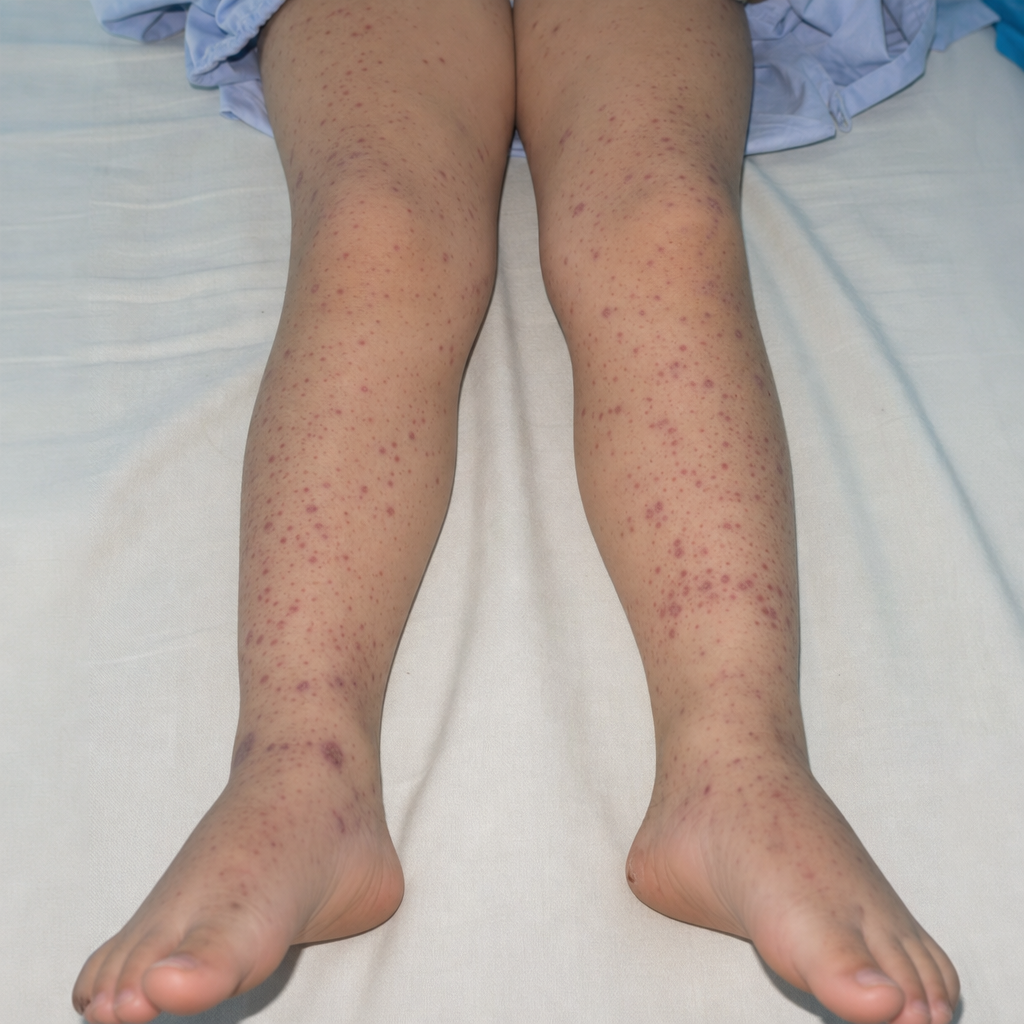
A 10 year old male child presents with purpuric rashes on the lower extremities, hematuria, abdominal pain, and arthritis but has no history of fever. What is the likely diagnosis ?
All are common features of juvenile idiopathic arthritis EXCEPT
Pauciarticular JRA is characterized by all except:
Henoch Schonlein purpura commonly involves which age group?
Which category of patients with juvenile idiopathic arthritis, who are mostly HLA-B27 positive and present with enthesitis, lower limb arthritis involving the knees and ankles, and inflammatory low back pain, are most likely to be?
Which of the following is NOT true about Henoch-Schönlein purpura?
Which of the following is not true about Juvenile rheumatoid arthritis?
A 5-year-old boy has recurrent infections, low platelet count, and rash as shown below. What is the likely diagnosis?
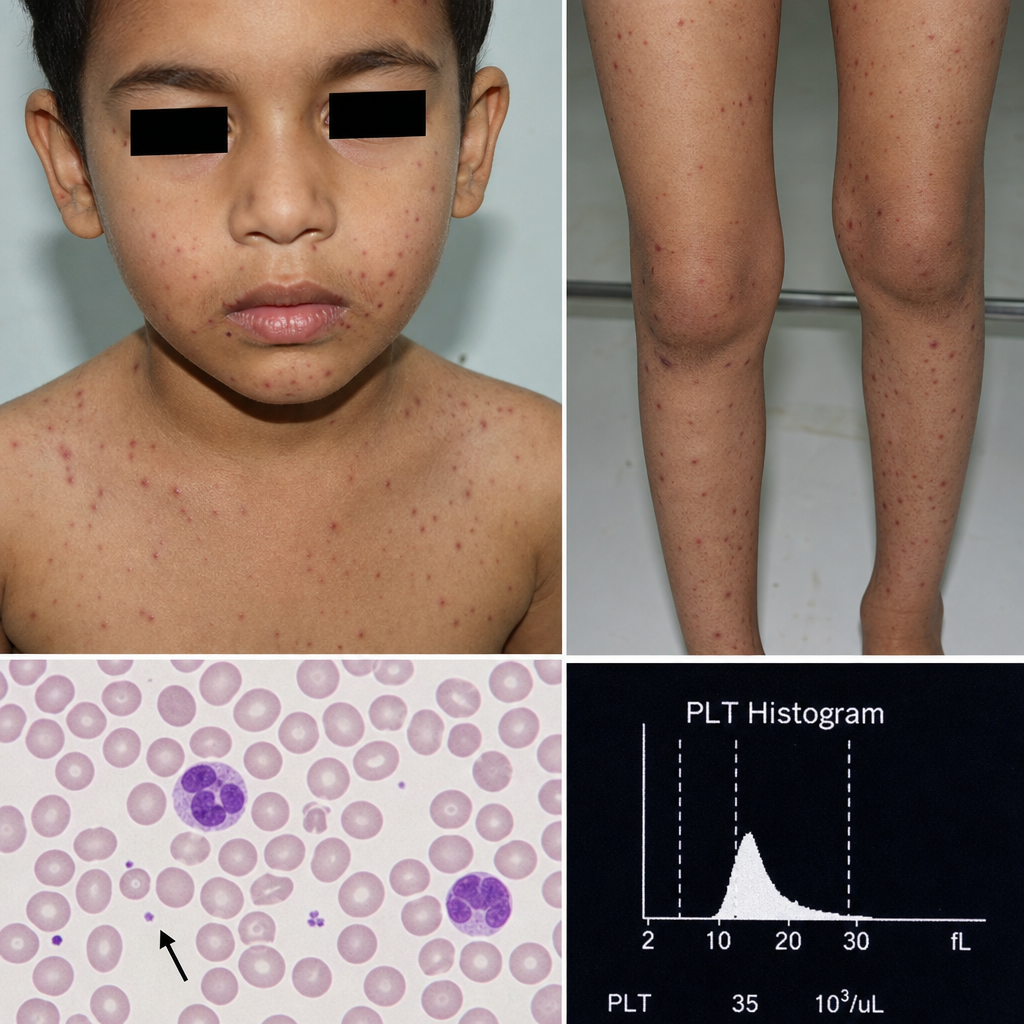
A 5 yr old male child presented with H/O recurrent infection. O/E the child has rashes as shown below. Routine blood investigation reveal patient has low platelets. Which of the following diagnosis is possible?
Practice by Chapter
Development of Immune System
Practice Questions
Primary Immunodeficiency Disorders
Practice Questions
Secondary Immunodeficiency Disorders
Practice Questions
Allergic Rhinitis
Practice Questions
Asthma in Children
Practice Questions
Atopic Dermatitis
Practice Questions
Food Allergies
Practice Questions
Drug Allergies
Practice Questions
Anaphylaxis
Practice Questions
Urticaria and Angioedema
Practice Questions
Autoimmune Disorders
Practice Questions
Immunotherapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app