
Immunology and Allergies — MCQs

On this page
A 2.5-year-old boy is brought to the nutrition clinic. He was born at term with a birth weight of 3.0 kg. His mother reports that since weaning he has been fed almost exclusively on a maize-based porridge with minimal animal protein or legumes. On examination, he weighs 9.8 kg and has a height of 82 cm. He has bilateral pitting oedema up to the ankles, generalised skin changes with areas of hyperpigmentation and hypopigmentation, and hair that shows alternating light and dark bands. Serum albumin is 1.8 g/dL. Haemoglobin is 8.2 g/dL. Stool microscopy shows no parasites. Which of the following biochemical profiles is most consistent with this child's nutritional disorder?
What is a characteristic feature of Systemic Juvenile Idiopathic Arthritis?
An 8-year-old child has had abdominal pain and dark urine for 10 days. Physical examination shows blotchy purple skin lesions on the trunk and extremities. Urinalysis shows hematuria and proteinuria. Serologic test results are negative for myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA) and proteinase 3-antineutrophil cytoplasmic antibody (PR3-ANCA). A skin biopsy specimen shows necrotizing vasculitis of small dermal vessels. A renal biopsy specimen shows immune complex deposition in glomeruli, with some IgA-rich immune complexes. Which of the following is the most likely diagnosis?
A child presents with delayed separation of the umbilical cord, leukocytosis, Down syndrome, and recurrent infections. What is the most likely diagnosis?
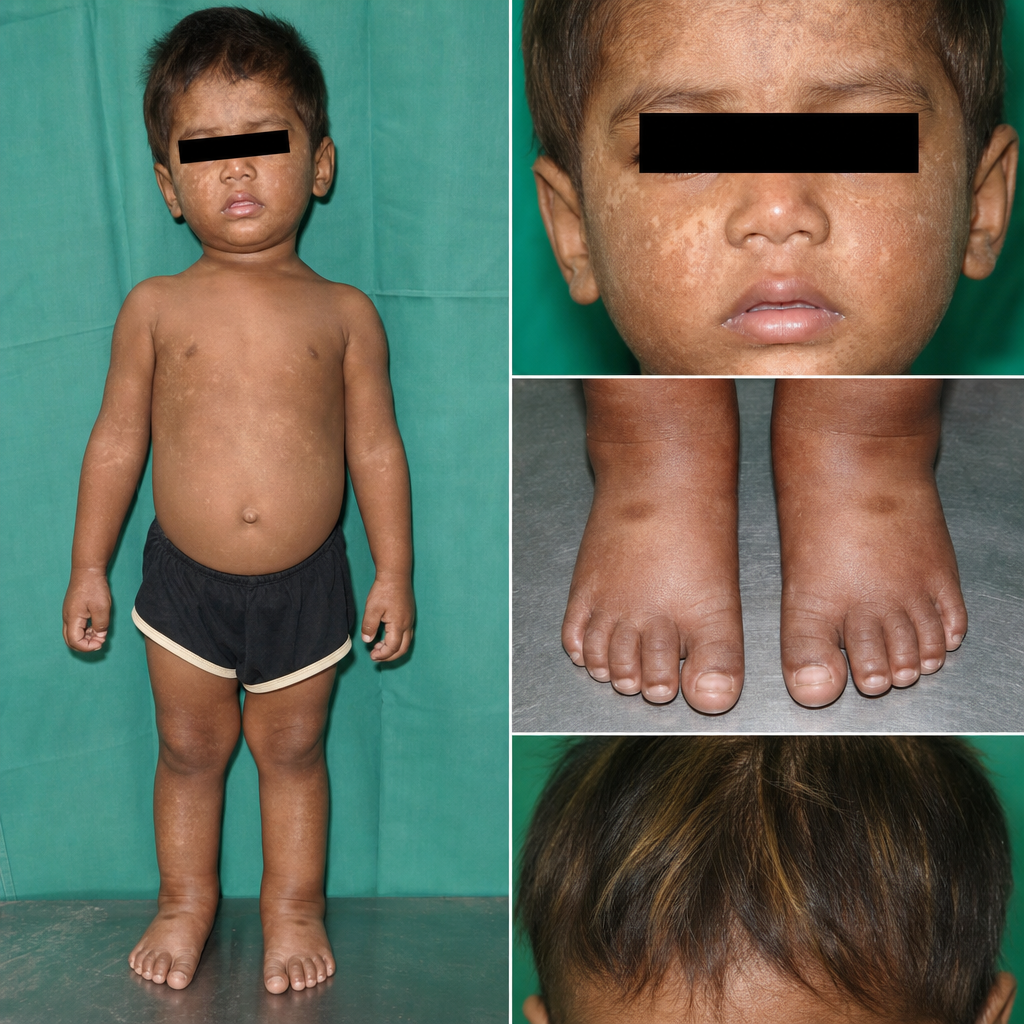
A 5-year-old male child presents with a history of recurrent infections and rashes, as shown in the image. Routine blood investigations reveal thrombocytopenia. Which of the following diagnoses is most likely?
Which of the following is NOT a criterion for Juvenile Rheumatoid Arthritis?
A child presents with a constellation of findings including fever, disabling arthritis, rash, and blindness. What is the MOST likely diagnosis?
Which vasculitis is seen most commonly in childhood?
Which of the following is diagnostic for juvenile idiopathic arthritis (JIA)?
A child has been diagnosed with cow milk allergy. Which of the following is the next recommended management strategy?
Practice by Chapter
Development of Immune System
Practice Questions
Primary Immunodeficiency Disorders
Practice Questions
Secondary Immunodeficiency Disorders
Practice Questions
Allergic Rhinitis
Practice Questions
Asthma in Children
Practice Questions
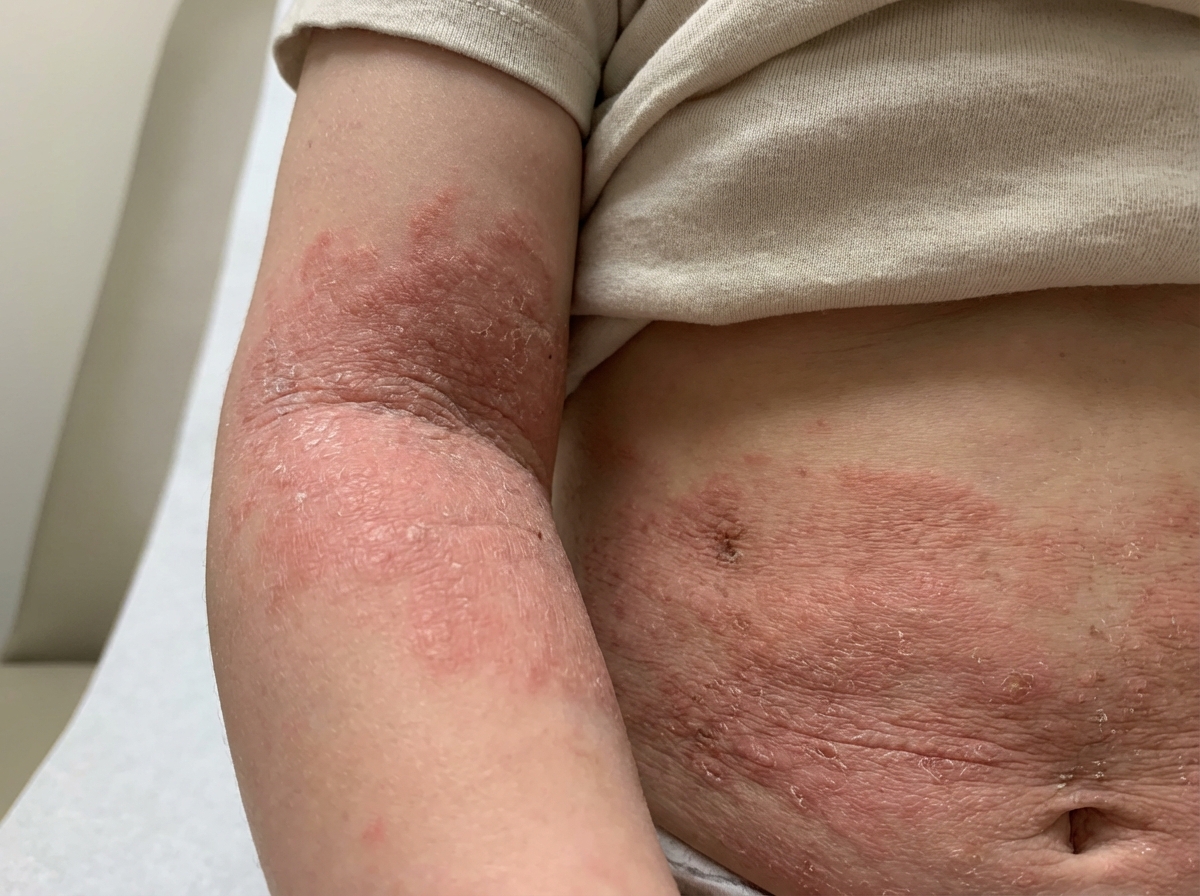
Atopic Dermatitis
Practice Questions
Food Allergies
Practice Questions
Drug Allergies
Practice Questions
Anaphylaxis
Practice Questions
Urticaria and Angioedema
Practice Questions
Autoimmune Disorders
Practice Questions
Immunotherapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app