
Hematology — MCQs

On this page
A nine-month-old boy presented with progressive lethargy, irritability, and pallor since 6 months of age. Examination revealed severe pallor. Investigations showed Hb 3.8 mg/dL, MCV 58 fL, MCH 19.4 pg/cell. Blood film shows normal osmotic fragility, target cells, and normoblasts. X-ray skull shows expansion of the erythroid marrow. Which of the following is the most likely diagnosis?
In infants, from which bone is a bone marrow biopsy typically performed?
An 18-month-old boy presented with a tender, swollen, warm right knee with significant hemarthrosis after an injury. His PT is 12 seconds (normal, 13 seconds), PTT is over 100 seconds (normal, 25 seconds), platelet count is 300,000/mm 3, and bleeding time is normal. What should be the initial management?
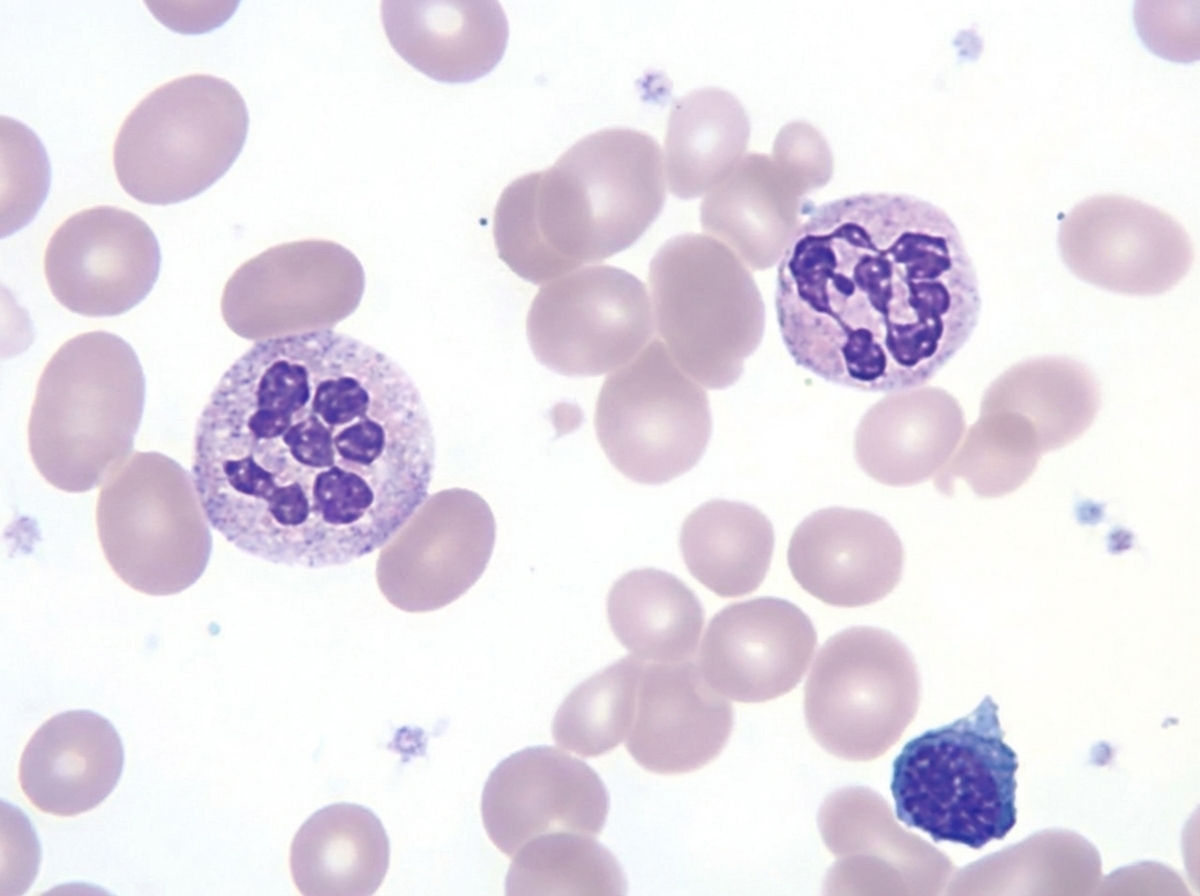
An otherwise healthy 1-year-old child has a routine CBC revealing polymorphonuclear neutrophils as shown below. Which of the following is the most appropriate next step?
What is true about Immune Thrombocytopenic purpura (ITP)?
A male baby was born in a hospital. After 12 hours of birth, the baby was found to be pale. His serum total bilirubin level was 20 mg/dl and unconjugated bilirubin was 15 mg/dl. After 36 hours of birth, Hemoglobin was 14 g/dl, and the reticulocyte count was high. On peripheral smear, nucleated RBCs and spherocytes are seen. What is the best possible diagnosis?
A child presents with bleeding gums and a swollen knee. What is the most likely underlying cause?
Constitutional pancytopenia can be seen in which of the following conditions, except?
Diamond Blackfan anemia is another name for which of the following conditions?
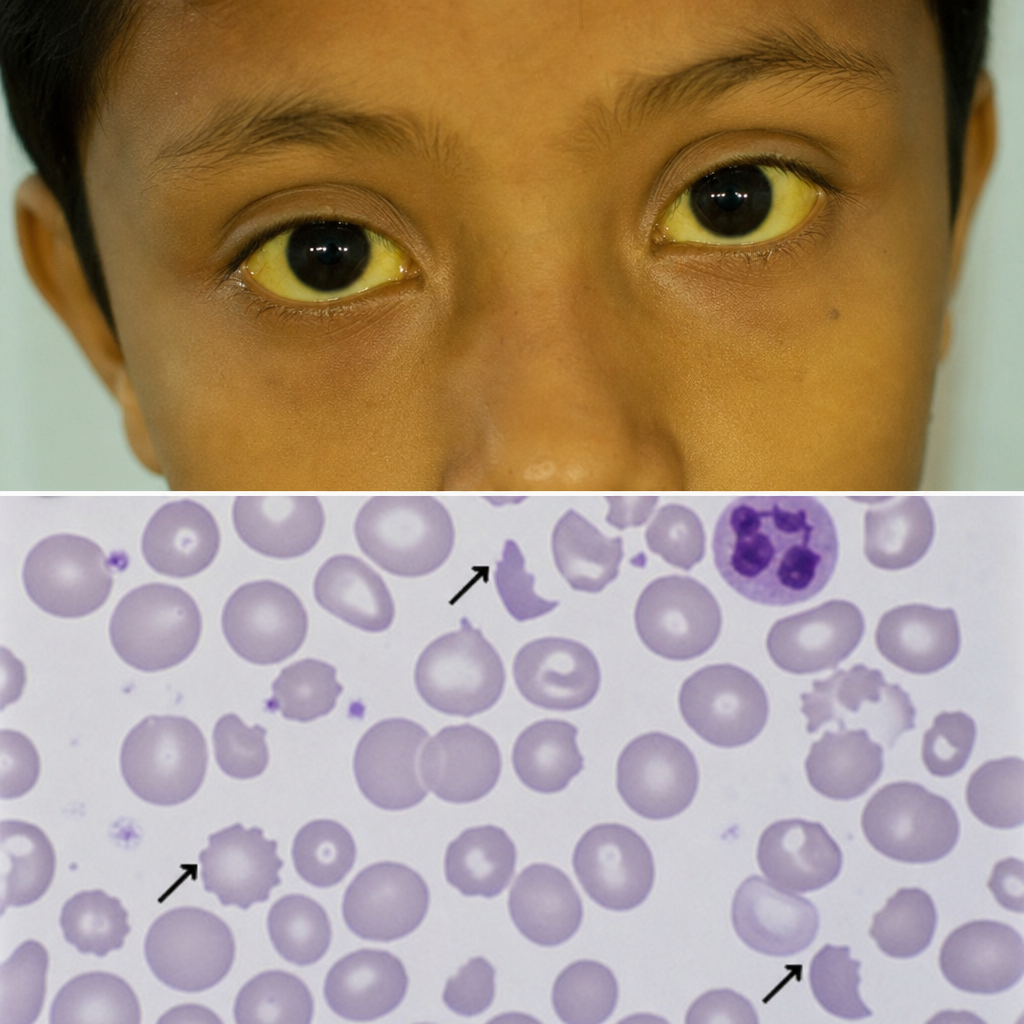
A 7-year-old child presented with high-grade fever with chills and rigor. Peripheral smear examination revealed Plasmodium vivax. The child was treated for malaria. Subsequently, the child presented with jaundice, and a repeat peripheral smear showed abnormalities. What is your diagnosis?
Practice by Chapter
Anemias in Children
Practice Questions
Hemoglobinopathies
Practice Questions
Hemolytic Anemias
Practice Questions
Nutritional Anemias
Practice Questions
Thrombocytopenia
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
White Blood Cell Disorders
Practice Questions
Bone Marrow Failure Syndromes
Practice Questions
Blood Component Therapy
Practice Questions
Hemophilia and Von Willebrand Disease
Practice Questions
Evaluation of Bleeding Tendencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app