
Hematology — MCQs

On this page
Which of the following is NOT related to fetal erythropoiesis?
A 5-year-old male child presents with severe transfusion-requiring anemia and jaundice. On examination, the liver and spleen are palpable 5 cm below the costal margin. Peripheral smear analysis shows findings consistent with a specific diagnosis. What is your diagnosis?
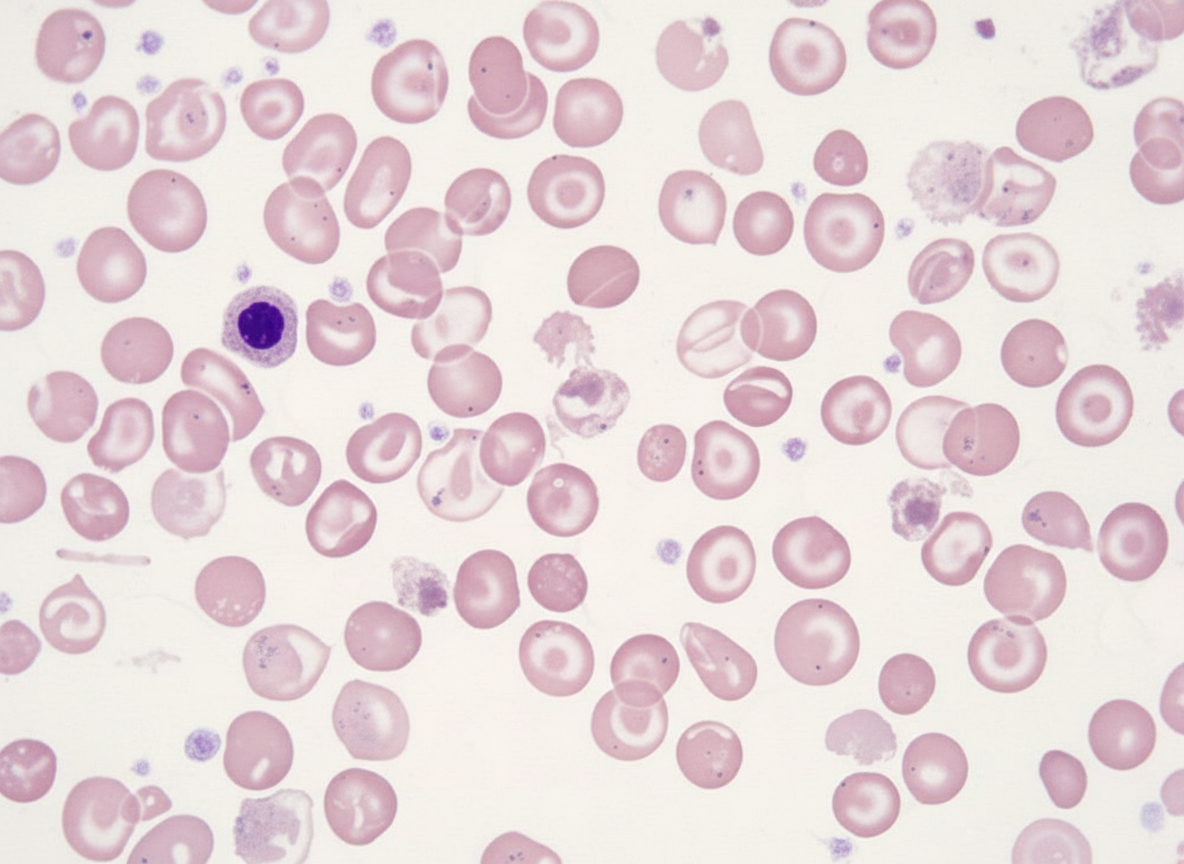
What is the first change of improvement noted after iron therapy is initiated?
A 7-year-old boy presented with sudden onset petechiae and purpura. There was a history of upper respiratory tract infection 2 weeks prior. On examination, there was no hepatosplenomegaly. What is the most probable diagnosis?
A 20-month-old female child presents for a routine check-up. A complete blood count (CBC) reveals moderate neutropenia, but the child appears healthy, eats well, and is within expected developmental parameters for her age and sex. Other blood count parameters are within the normal range for her age. The family history is unremarkable. Repeat CBCs after one and two weeks show persistent neutropenia. Bone marrow examination is normal. What is the next best step in management?
Macrocytic anemia in children is produced by all of the following except?
A 5-year-old boy presents with petechial spots that appeared overnight. Two weeks prior, he experienced abdominal pain. There is no hepatosplenomegaly. What is the most likely diagnosis?
A 6-week-old child presents with a hemoglobin of 10 gm% and appears pale on examination. What is the most likely diagnosis?
Frameshift mutation occurs due to:
A 2-year-old child presents with short stature and cafe-au-lait spots. Bone marrow aspiration yields a little material and mostly containing fat. What is your diagnosis?
Practice by Chapter
Anemias in Children
Practice Questions
Hemoglobinopathies
Practice Questions
Hemolytic Anemias
Practice Questions
Nutritional Anemias
Practice Questions
Thrombocytopenia
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
White Blood Cell Disorders
Practice Questions
Bone Marrow Failure Syndromes
Practice Questions
Blood Component Therapy
Practice Questions
Hemophilia and Von Willebrand Disease
Practice Questions
Evaluation of Bleeding Tendencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app