
Hematology — MCQs

On this page
A 7-year-old boy has severe microcytic anemia due to beta-thalassemia major (homozygous). He requires frequent blood transfusions to prevent skeletal and developmental complications. Which of the following medications is also indicated in the treatment of patients requiring frequent blood transfusions?
A 7-year-old child is brought to the clinic with complaints of fatigue and poor concentration. The mother reports that the child has been eating non-food items such as chalk and soil for the past few months. A peripheral blood smear image shows microcytic, hypochromic red blood cells. Which of the following is the most likely diagnosis?
A child with progressive pallor and bone pain has an elevated HbS based on the HPLC report. Which is the best treatment to manage hemolysis in this patient?
A young boy presented with petechiae and his platelet count was 10,000/cu mm. Bone marrow aspirate revealed normal cellularity with megakaryocyte hyperplasia. Most appropriate initial therapy?
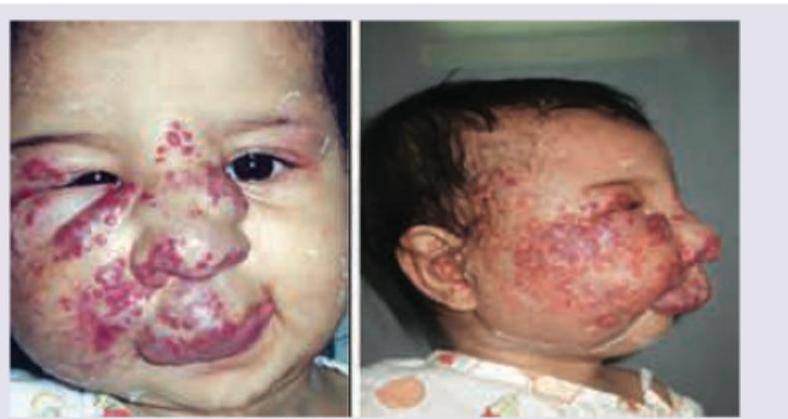
A 1-year-old infant with the following lesion on face. CNS examination was normal. Blood counts show thrombocytopenia with P. smear suggestive of microangiopathic changes. Probable diagnosis is:
A 6-month-old child brought by parents for diffuse ecchymosis on extremities and trunk. Probable diagnosis is? (Recent NEET Pattern 2016-17)
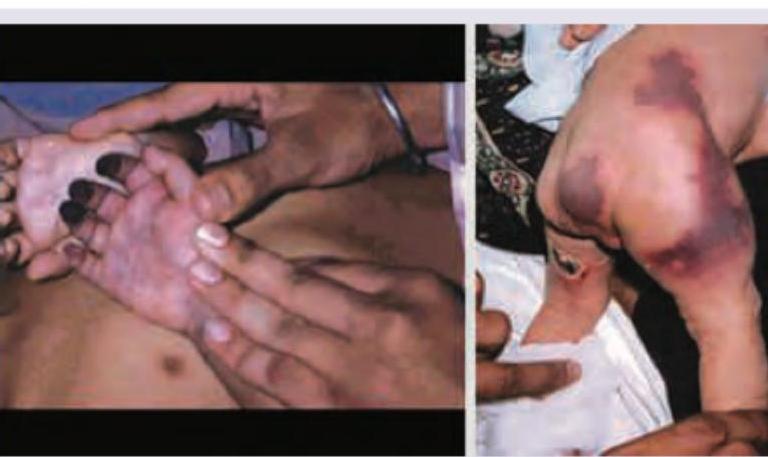
A 3-year-old child presents with sudden onset generalized petechiae and bruise on forehead. Sternal tenderness is absent and liver and spleen are not palpable. Bone marrow aspiration is normal. Probable cause is? (Recent NEET Pattern 2016-17)
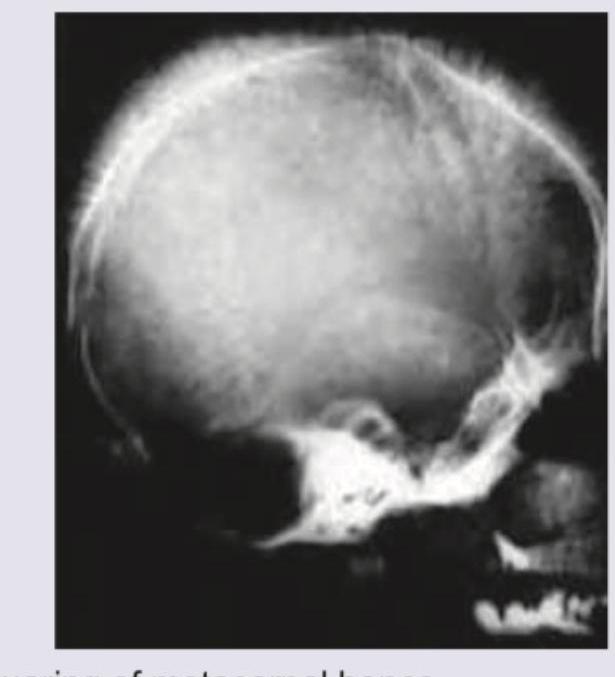
An 8-year-old boy with requirement of multiple blood transfusions. X-ray skull was performed. All are true about the condition shown except:
A 12-year-old child presents with acute kidney injury after a bout of dysentery. Not seen is:
A 10-year-old boy presented with fatigue. Investigations revealed: Hemoglobin, 9 g/dL; MCV, 60 fL; MCH, 20 pg; and serum ferritin, 185 µg/L. The TLC was elevated and showed predominant lymphocytes and neutrophils. What is the likely diagnosis in this patient? **Normal values:** - Serum ferritin: 50-150 µg/L
Practice by Chapter
Anemias in Children
Practice Questions
Hemoglobinopathies
Practice Questions
Hemolytic Anemias
Practice Questions
Nutritional Anemias
Practice Questions
Thrombocytopenia
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
White Blood Cell Disorders
Practice Questions
Bone Marrow Failure Syndromes
Practice Questions
Blood Component Therapy
Practice Questions
Hemophilia and Von Willebrand Disease
Practice Questions
Evaluation of Bleeding Tendencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app