
Bone Marrow Failure Syndromes — MCQs

What is the best treatment option for a patient aged 65 years with severe aplastic anemia who has an HLA-compatible sibling available?
An 18-year-old male presents to the OPD with gum bleeding, fever, low total leukocyte count (TLC), and low platelet count. General examination is unremarkable. Further investigations reveal a low reticulocyte count, absent megakaryocytes, and no immature cells in the bone marrow. What is the most likely diagnosis?
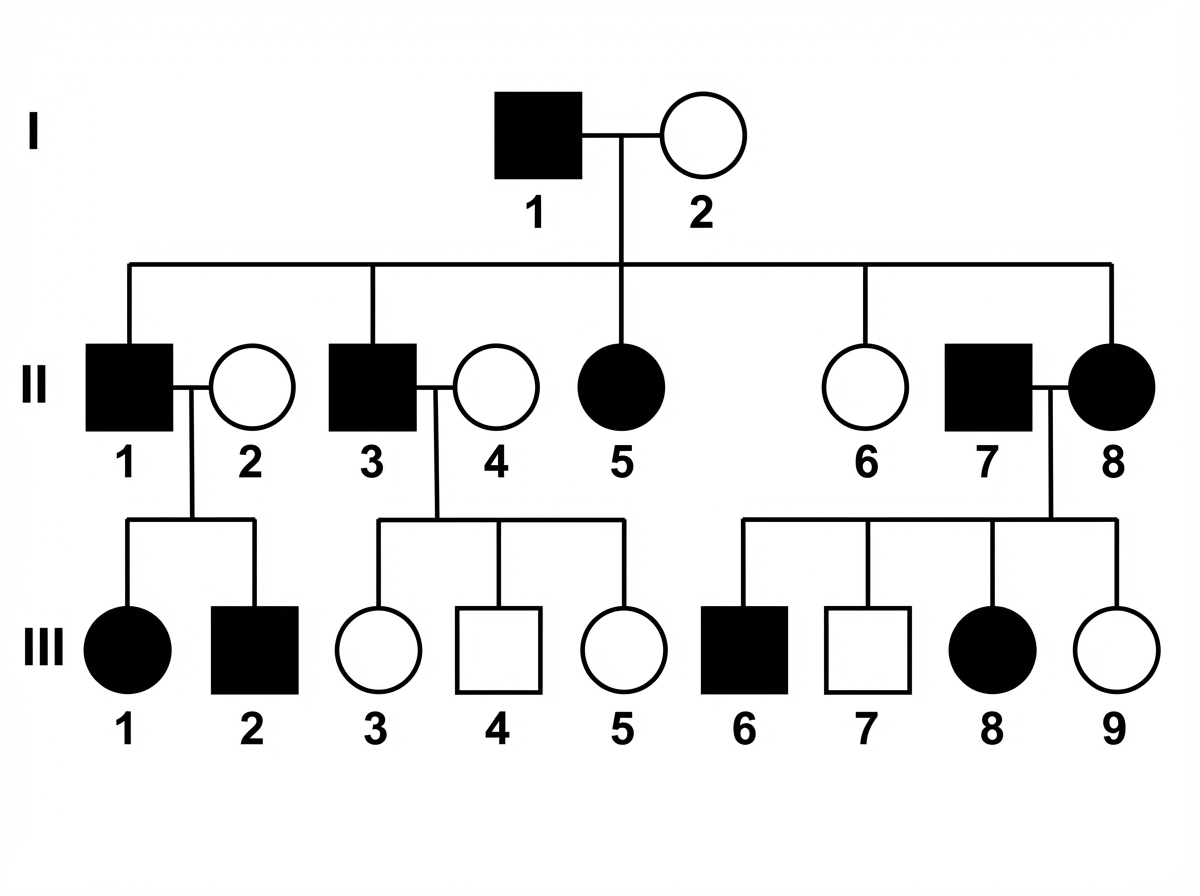
Which disease will show the mode of inheritance depicted in this pedigree?
A 69-year-old woman, with poor dietary habits and alcoholism, is found to have a macrocytic anemia with hyper segmented neutrophils. Which of the following is the most appropriate diagnostic test?
Which of the following is not a characteristic of Fanconi's anemia?
False about Shwachman-Diamond syndrome
All are associated with malignancy except
Which of the following is not a chromosome breakage disorder?
The strongest occupational risk factor for hematological carcinoma is
Which of the following is most specific for congenital Rubella syndrome?
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app