
Growth and Development — MCQs

On this page
A 6-year-old child is brought for complaints of short stature. His height is comparable with his parent's height and his bone age corresponds to his chronological age. What is the likely diagnosis?
Gonadal growth corresponds with _____.
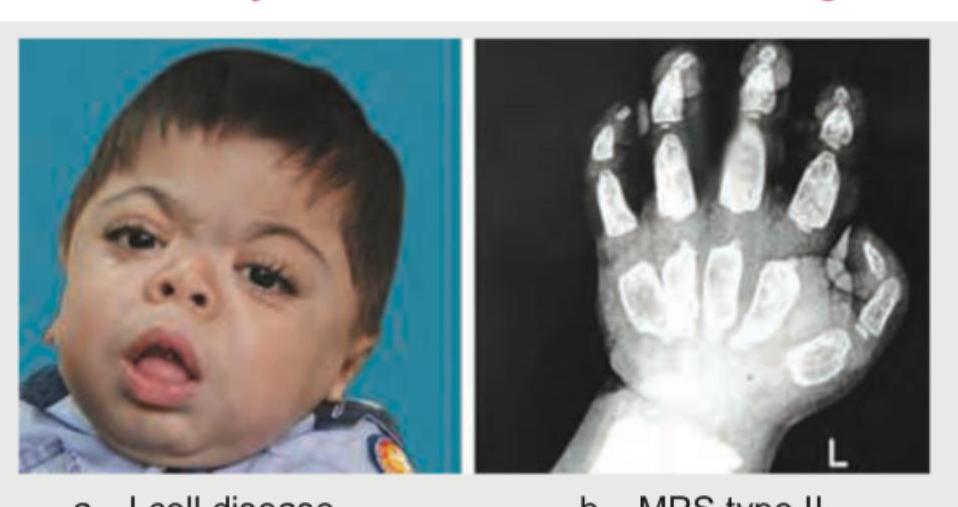
A 10-month-old child with coarse facies is referred for developmental delay. On examination, hepatosplenomegaly was noted. WBC N-acetylglucosamine-1-phosphotransferase activity was absent. The X-ray is shown below. What is the diagnosis?
A mother is concerned about the bowing of legs of her child. Which tests should be performed to label it as physiological genu varum?
A 6-year-old child is brought with the following lesion. He can develop lesions in which organ in future?
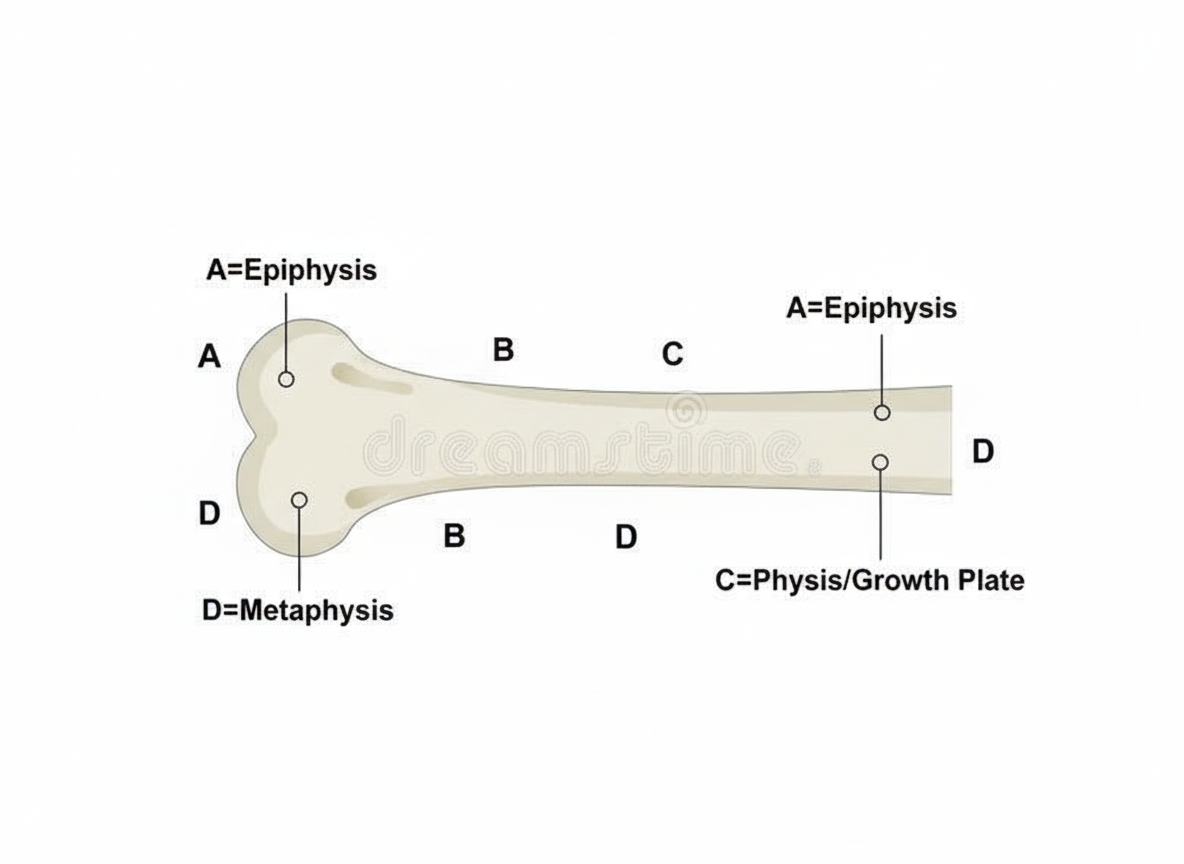
Which structure is most commonly affected in rickets?
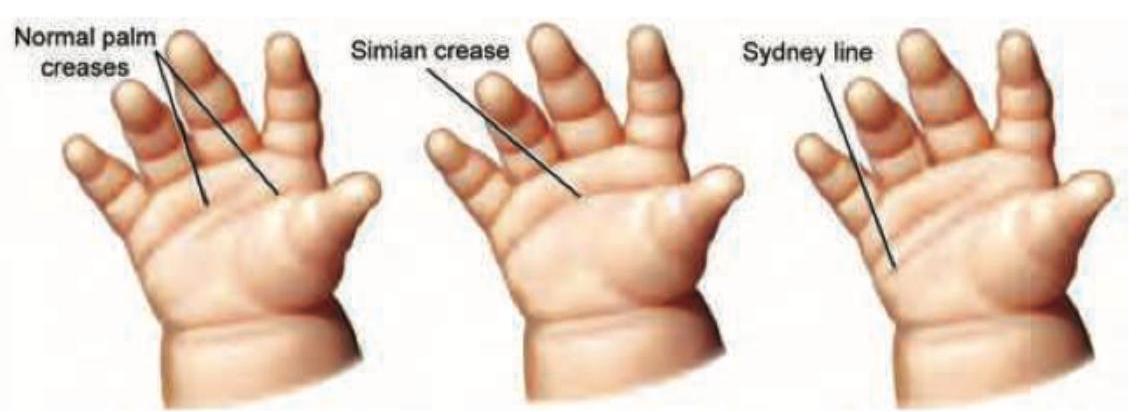
The image displays a hand with a distinct palmar crease pattern where a transverse crease extends from the radial to the ulnar border, in addition to another proximal transverse crease. What is this specific crease pattern called?
The picture depicts Rocker bottom feet. This finding is classically associated with which chromosomal abnormality?
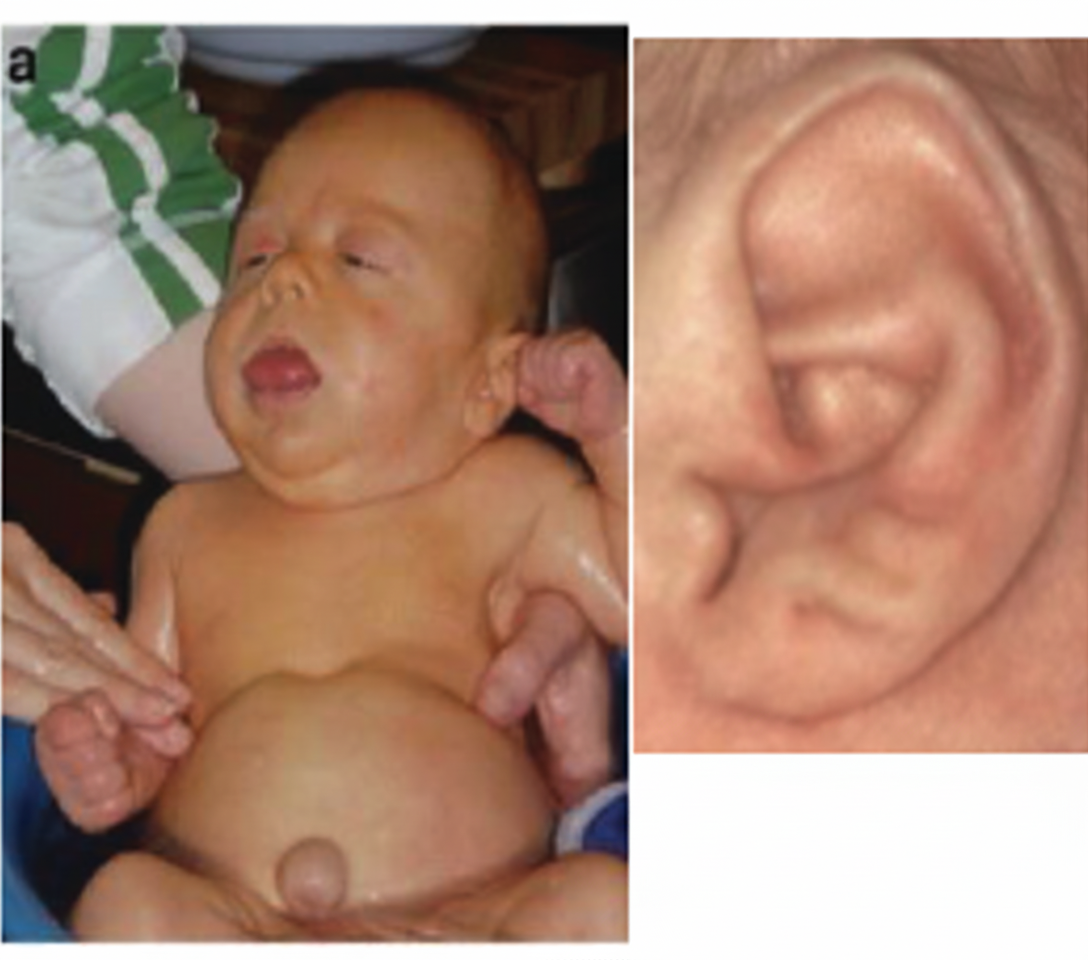
Ear lobe creases in a child are commonly associated with which disorder that also presents with macroglossia and macrosomia?
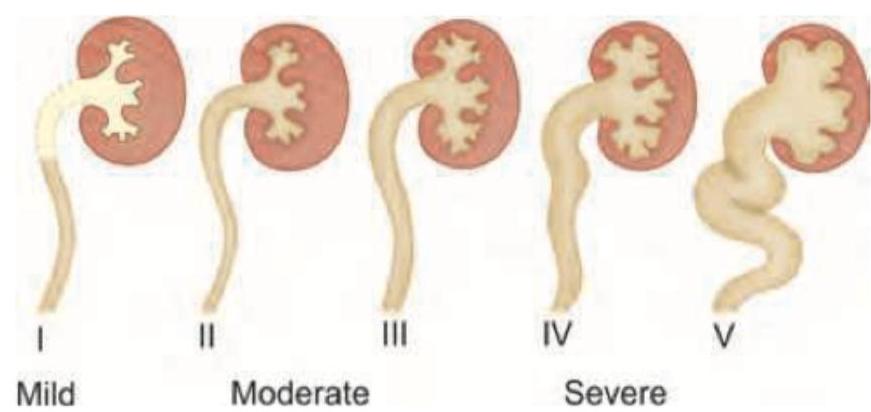
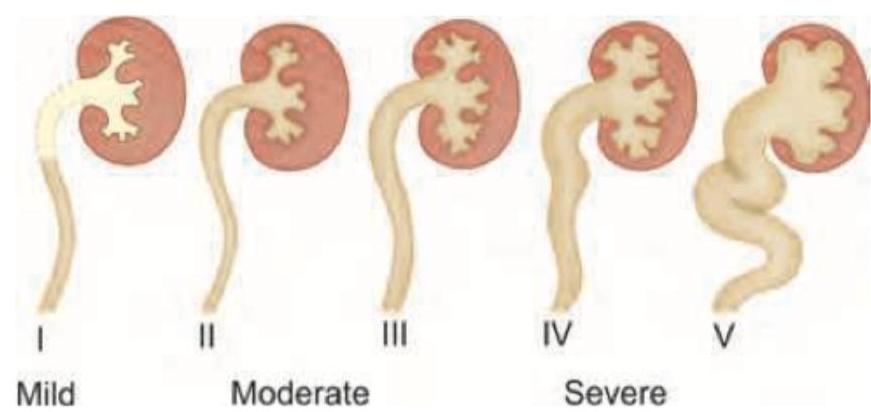
A child presents with polydactyly and obesity. Which syndrome is most likely associated with these clinical findings?
Practice by Chapter
Normal Growth Parameters
Practice Questions
Developmental Milestones
Practice Questions
Puberty and Adolescent Development
Practice Questions
Growth Disorders
Practice Questions
Failure to Thrive
Practice Questions
Developmental Screening and Assessment
Practice Questions
Developmental Delays
Practice Questions
Growth Charts and Monitoring
Practice Questions
Short Stature
Practice Questions
Tall Stature
Practice Questions
Precocious and Delayed Puberty
Practice Questions
Psychosocial Development
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app