
Growth and Development — MCQs

On this page
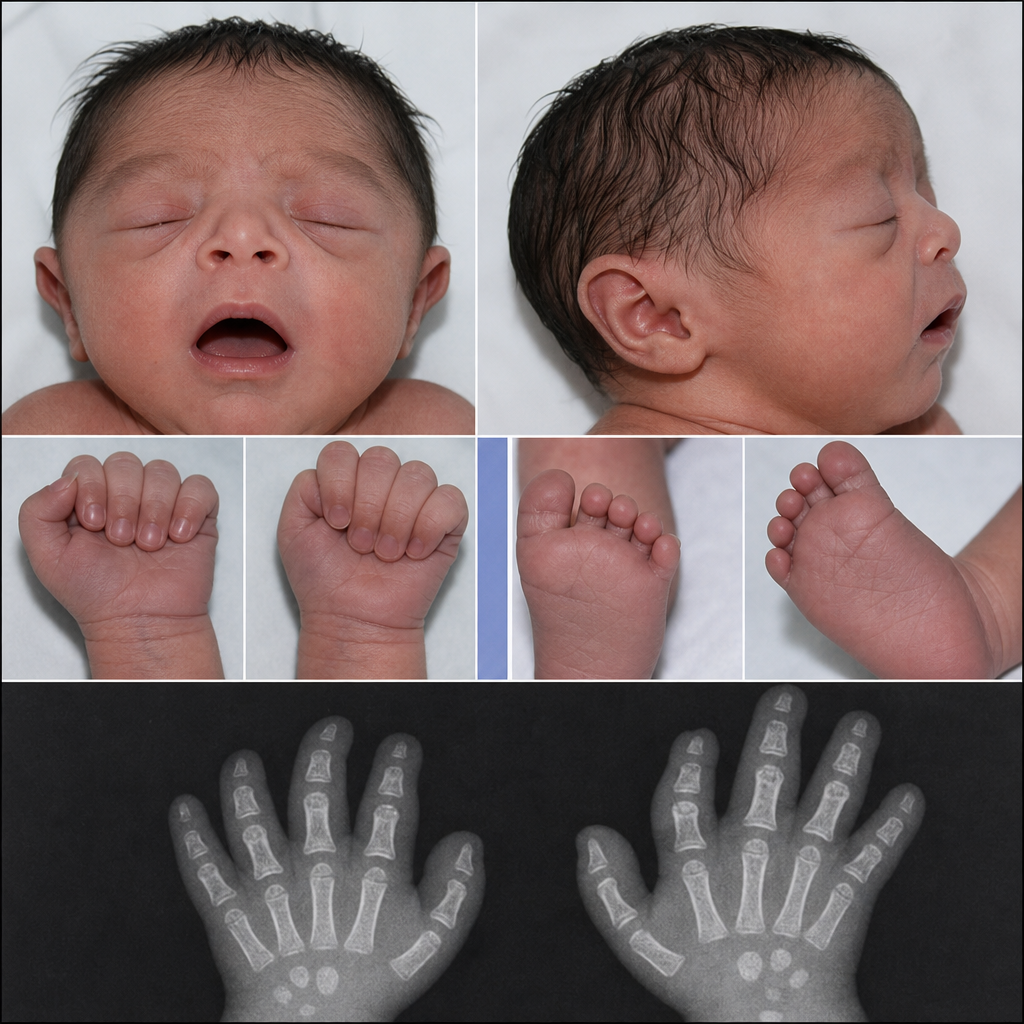
A neonate presented to OPD with specific clinical features. What is your diagnosis?
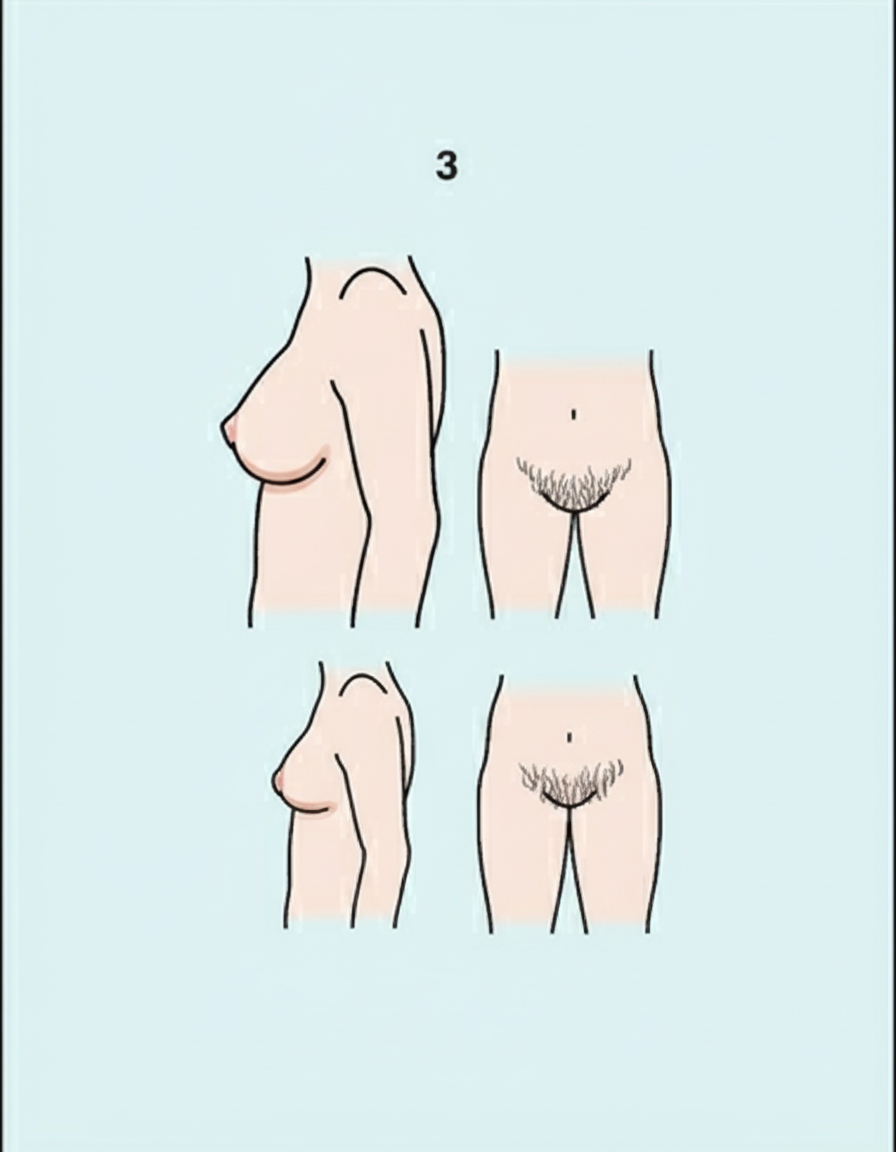
What is the Tanner-Whitehouse (SMR) staging based on the following findings in a female?
In Down's syndrome, what is the characteristic shape of the head?
What is the following device used to measure?

You are examining an infant and the findings are as follows: Adductor angle - 100°, Popliteal angle - 90°, Dorsiflexion angle of foot - 70°, Scarf sign - Elbow crosses the middle but doesn't reach the anterior axillary line. What is the appropriate age of the infant?
Tears are produced in the newborn after which period?
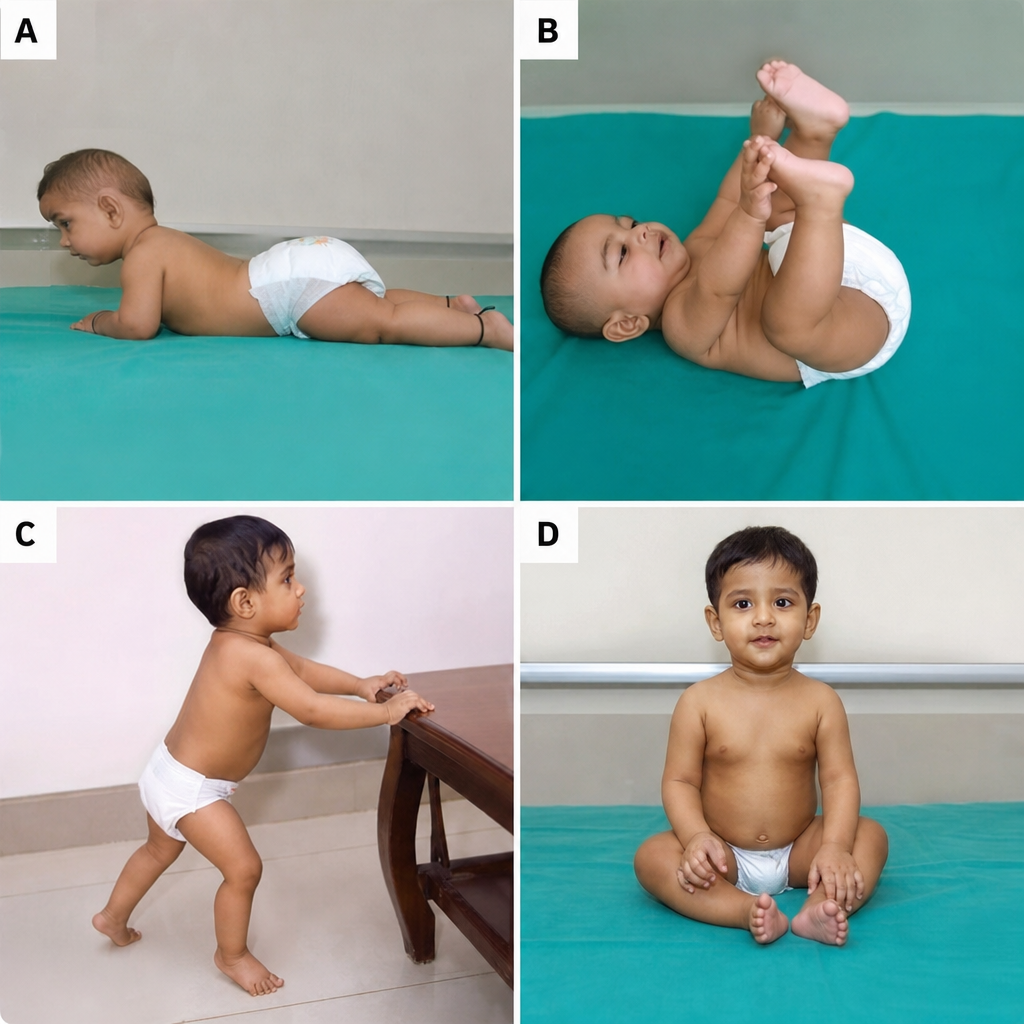
Which sequence represents the order in which the following milestones are typically attained?
Which of the following is true about Down's syndrome?
A child born normal, at 1 year of age, presents with an enlarged tongue, coarse facies, prominent forehead, flat face, depressed nasal bridge, and hepatosplenomegaly. What is the likely diagnosis?
A normal child typically achieves complete neck control by what age?
Practice by Chapter
Normal Growth Parameters
Practice Questions
Developmental Milestones
Practice Questions
Puberty and Adolescent Development
Practice Questions
Growth Disorders
Practice Questions
Failure to Thrive
Practice Questions
Developmental Screening and Assessment
Practice Questions
Developmental Delays
Practice Questions
Growth Charts and Monitoring
Practice Questions
Short Stature
Practice Questions
Tall Stature
Practice Questions
Precocious and Delayed Puberty
Practice Questions
Psychosocial Development
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app