
Gastroenterology — MCQs

On this page
In congenital pyloric stenosis, which of the following is NOT typically seen?
A 14 kg child has severe diarrhea for 6 hours. What is the recommended fluid replacement?
Most common cause of portal hypertension in children is _______?
What is the concentration of sodium in low osmolar ORS?
What is the most characteristic feature of congenital hypertrophic pyloric stenosis?
A 4-year-old girl presents with severe vomiting after a viral fever of 6 days and subsequently develops cerebral edema. What would be the expected liver biopsy finding?
All are seen in Reye's syndrome except?
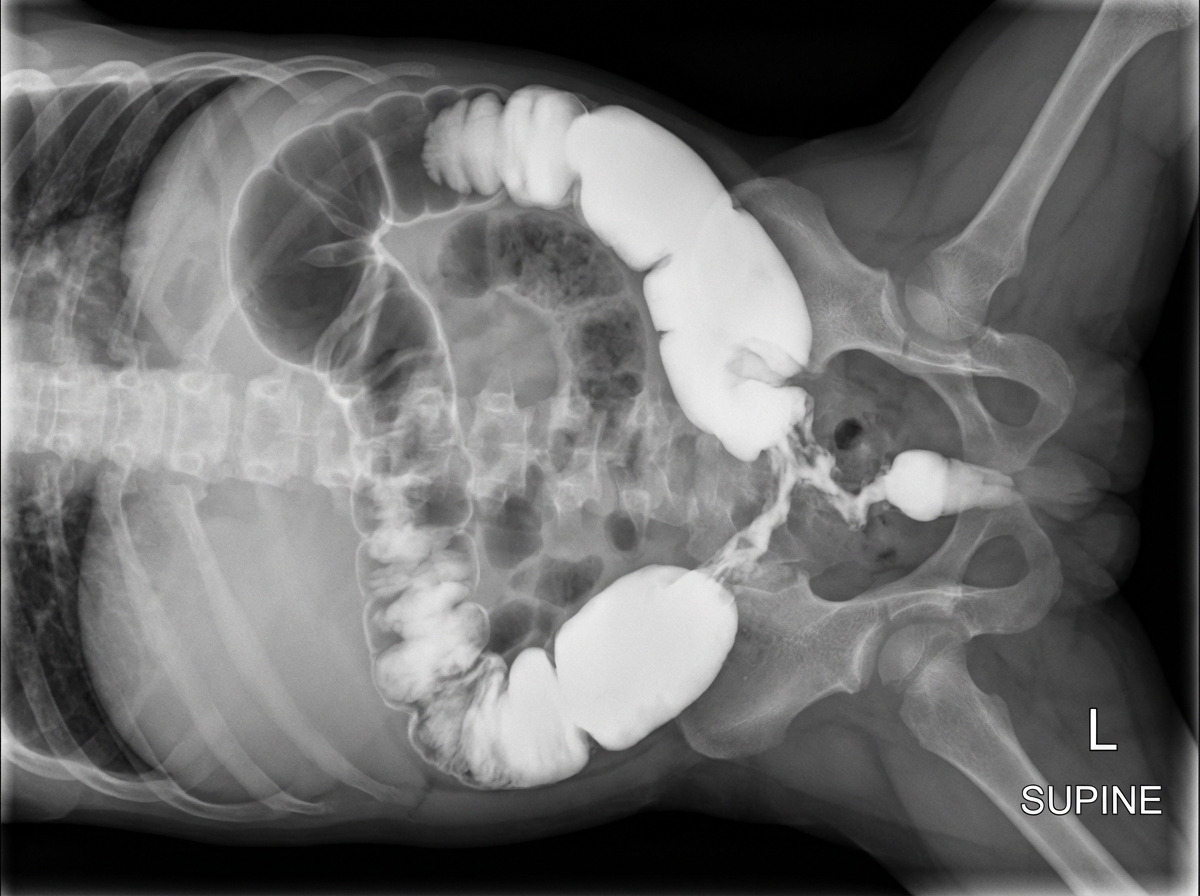
A 5-day-old term male neonate presents with delayed passage of meconium, abdominal distension, and bilious vomiting. Barium enema and intestinal biopsy findings are shown. What is the diagnosis?
What is the most common genetic cause of liver disease in children?
Which degree of dehydration is indicated by thirst in a child with diarrhea?
Practice by Chapter
Gastroesophageal Reflux
Practice Questions
Peptic Ulcer Disease
Practice Questions
Inflammatory Bowel Disease
Practice Questions
Celiac Disease
Practice Questions
Malabsorption Syndromes
Practice Questions
Acute and Chronic Diarrhea
Practice Questions
Constipation and Encopresis
Practice Questions
Gastrointestinal Bleeding
Practice Questions
Intestinal Obstruction
Practice Questions
Liver Diseases in Children
Practice Questions
Pancreatic Disorders
Practice Questions
Pediatric Nutritional Support
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app