
Endocrinology — MCQs

On this page
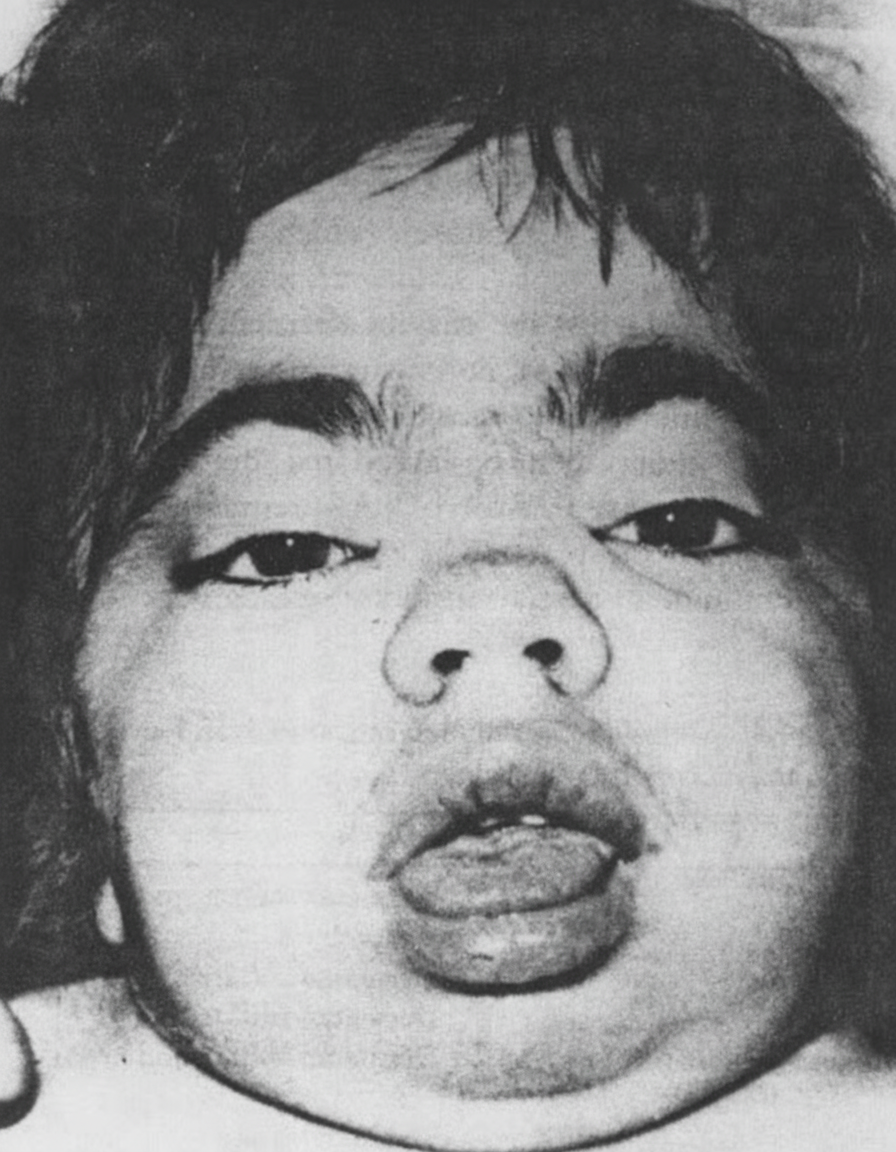
What is the disease this child, who also has hepatosplenomegaly and intellectual disability, is suffering from?
Which of the following is a common presentation of juvenile hypothyroidism?
A 2-week-old infant presents with protracted vomiting and poor oral intake. Examination reveals signs of dehydration and ambiguous genitalia. Serum electrolytes show hyponatremia, hypochloremia, and normal bicarbonate. Which of the following is the most common cause of ambiguous genitalia in this context?
The features of neonatal hyperthyroidism include all except?
Features of Laurence-Moon-Bardet-Biedl syndrome include all of the following EXCEPT?
A 2-year-old child presents with a history of drowsiness, progressing to unconsciousness and seizures. On evaluation, the blood glucose level is found to be 25 mg/dL. Following the administration of 5 mL/kg of dextrose, there is no improvement. A repeat blood glucose measurement shows the level has risen to 130 mg/dL. Considering this clinical scenario, which of the following interventions could be detrimental?
A newborn baby presents with tachycardia, hypotension, and irritability. USG revealed normal ovaries, and the karyotype is 46,XX. Which of the following biochemical abnormalities is not typically seen in this clinical scenario?
Inheritance pattern of Familial hypophosphatemic Rickets is:
Ambiguous genitalia in males is due to which of the following conditions?
The parents of a 4-week-old girl complain that their baby is apathetic and sluggish. On physical examination, the child's abdomen is large and exhibits an umbilical hernia. The skin is pale and cold, and the temperature is 35°C (95°F). Which of the following provides a plausible explanation for the signs and symptoms of this child?
Practice by Chapter
Disorders of Growth
Practice Questions
Thyroid Disorders
Practice Questions
Disorders of Puberty
Practice Questions
Adrenal Disorders
Practice Questions
Diabetes Mellitus in Children
Practice Questions
Disorders of Calcium and Phosphate Metabolism
Practice Questions
Disorders of Sexual Development
Practice Questions
Hypoglycemia
Practice Questions
Obesity and Metabolic Syndrome
Practice Questions
Pituitary Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Endocrine Emergencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app