
Endocrinology — MCQs

On this page
Serum glucose level in children > 2 months defining hypoglycemia is?
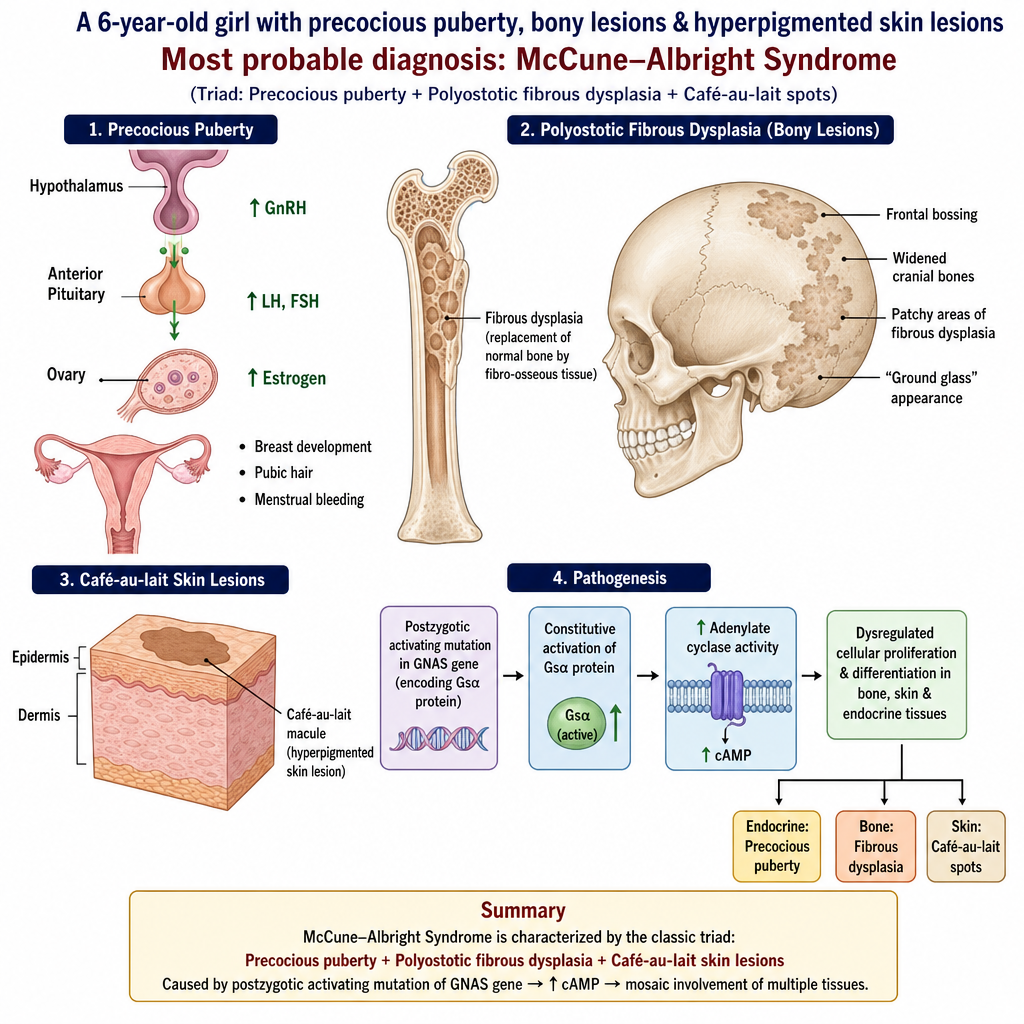
A 6-year-old girl presents with precocious puberty, some bony lesions & hyperpigmented skin lesions as shown below. What is the most probable diagnosis?
A 10 year old boy is having polyuria, polydipsia, laboratory data showed (in mEq/lit) – Na+ 154, K+ 4.5, HCO3- 22, Serum osmolality – 295 mOsm/kg, Blood urea – 50 mg/dl, Urine specific gravity – 1.005. The likely diagnosis is –
Which is false in Congenital Hypopituitarism?
What percentage of the total daily dose is given as basal insulin in a basal-bolus regimen in children?
A 6-year-old girl presents with a left breast mass. Her mother first noticed it a day before and is very concerned because both the child's maternal grandmother and maternal aunt have had breast cancer. It is firm, smoothly circumscribed, and slightly eccentric under the left areola. The right breast is unremarkable. You suggest:
All of the following may be causes of precocious puberty in girls, except:
The commonest cause of congenital hypothyroidism is -
Which of the following statements are True or False about Cretinism? 1. Goitre present at birth 2. Can be diagnosed by serum T4 levels 3. Most commonly seen in Iodine deficiency endemic areas 4. Skeletal development is delayed 5. Physiological jaundice is prolonged
Clinical features of "hypothyroidism" in a newborn are all except:
Practice by Chapter
Disorders of Growth
Practice Questions
Thyroid Disorders
Practice Questions
Disorders of Puberty
Practice Questions
Adrenal Disorders
Practice Questions
Diabetes Mellitus in Children
Practice Questions
Disorders of Calcium and Phosphate Metabolism
Practice Questions
Disorders of Sexual Development
Practice Questions
Hypoglycemia
Practice Questions
Obesity and Metabolic Syndrome
Practice Questions
Pituitary Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Endocrine Emergencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app