
Endocrinology — MCQs

On this page
These two brothers age 10 and 8 years are having short stature with midface hypoplasia, high squeaky voice. The levels of GH are elevated with normal TSH, FT4 and FT3. Diagnosis is?
The child in the image most likely suffers from?
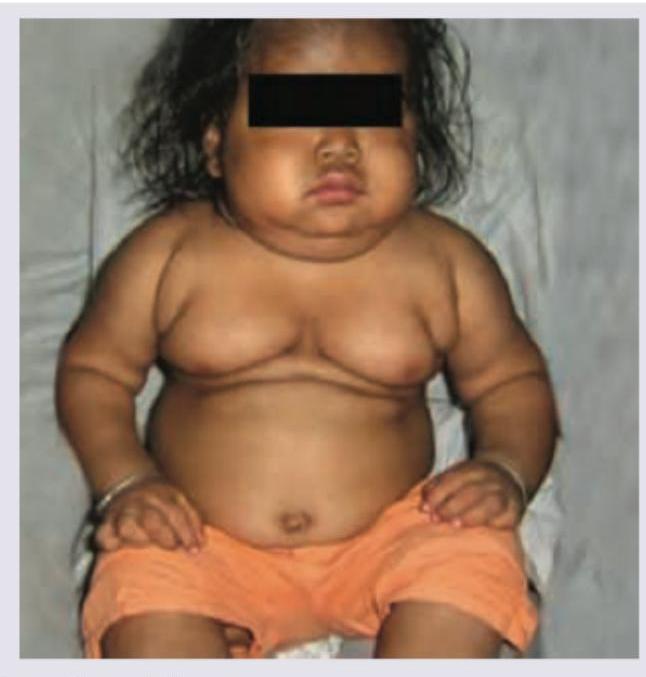
The following child with Genu Valgum has been treated with multiple dosages of injectable vitamin D3 supplements at a P.H.C. The lab values ordered by you shows serum calcium = 9 mg% with low serum phosphate and normal levels of 25-hydroxycholecalciferol. The most probable defect is?
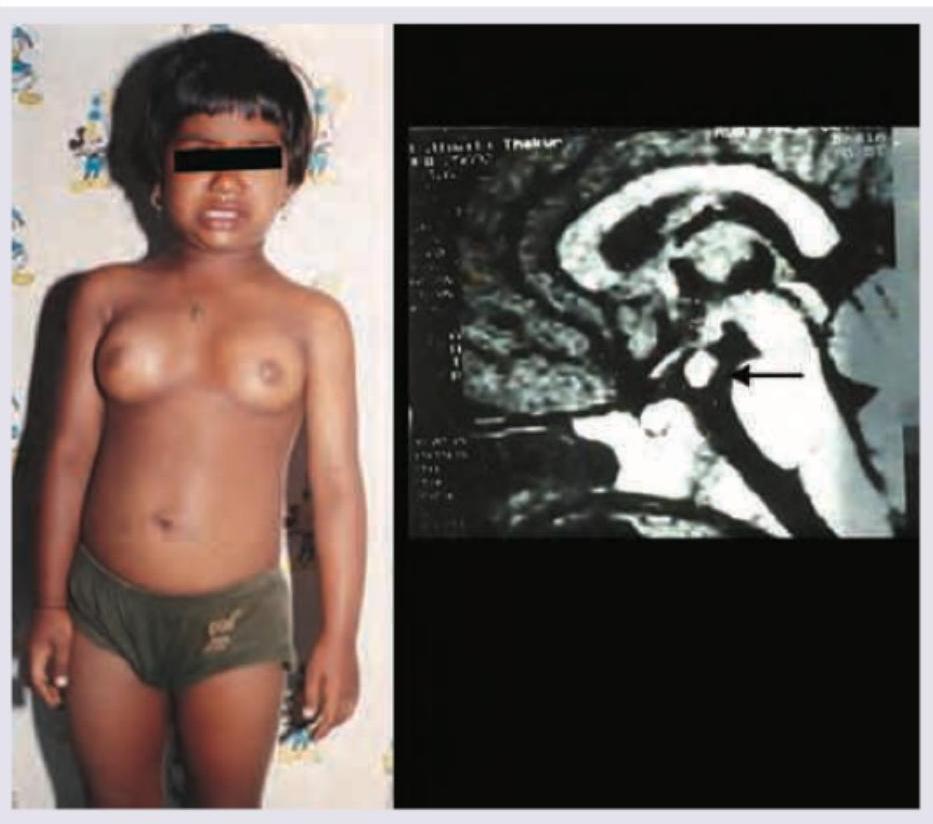
What is the most likely diagnosis based on the clinical presentation and MRI findings shown?
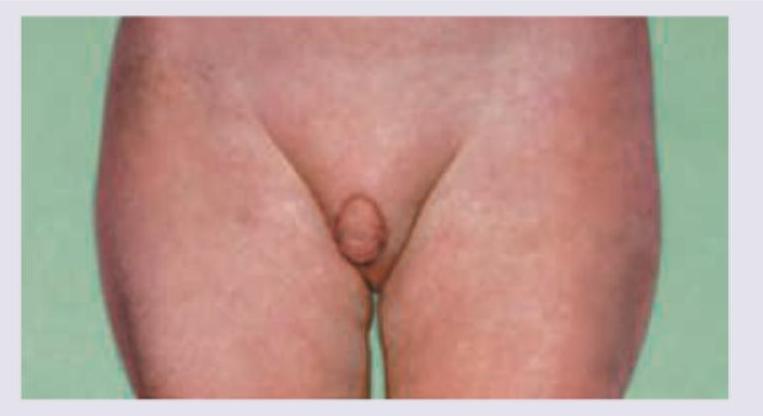
In this case of ambiguous genitalia, all of the following investigations are needed EXCEPT:
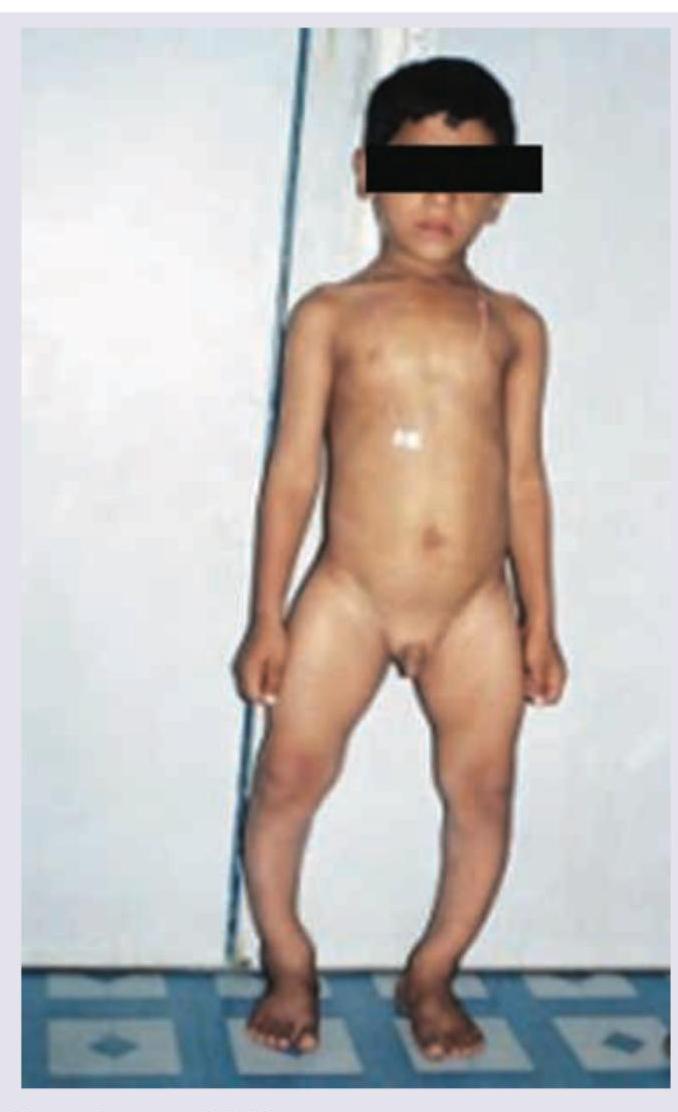
A 10-year-old short stature child presents with daily headache, vomiting, increased urine output and polydipsia. CT head is performed. All are true about the condition except:
All are seen in this girl child except:
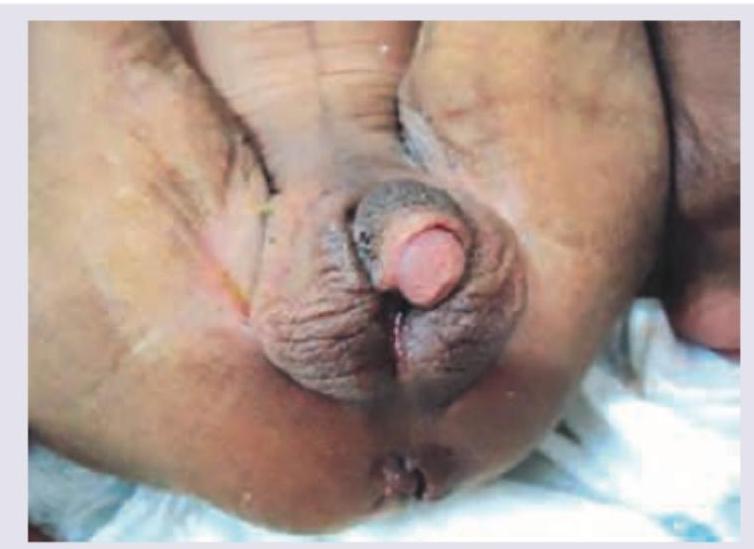
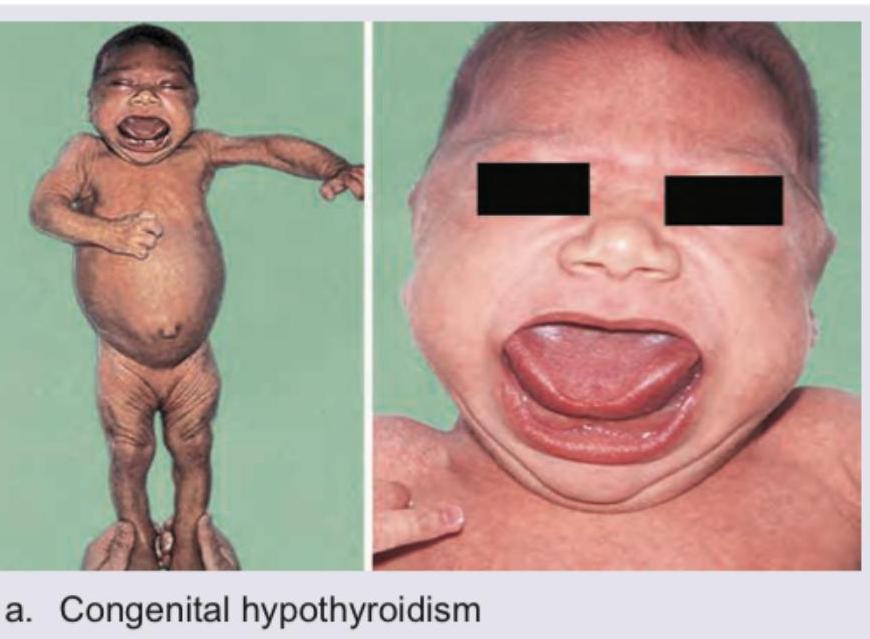
A neonate presents with the clinical features shown in the image below. What is the most likely diagnosis?
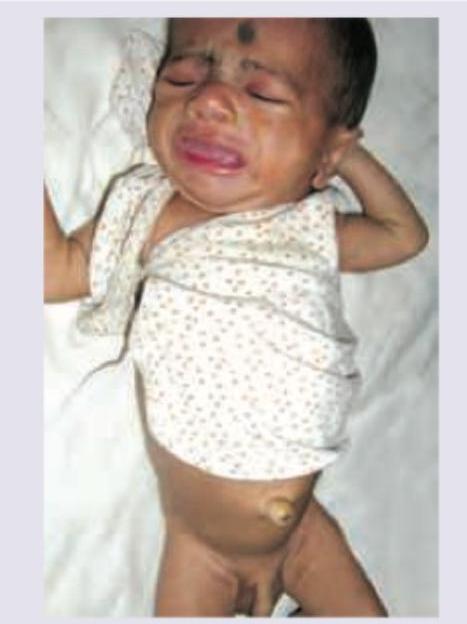
Comment on the diagnosis of the child:
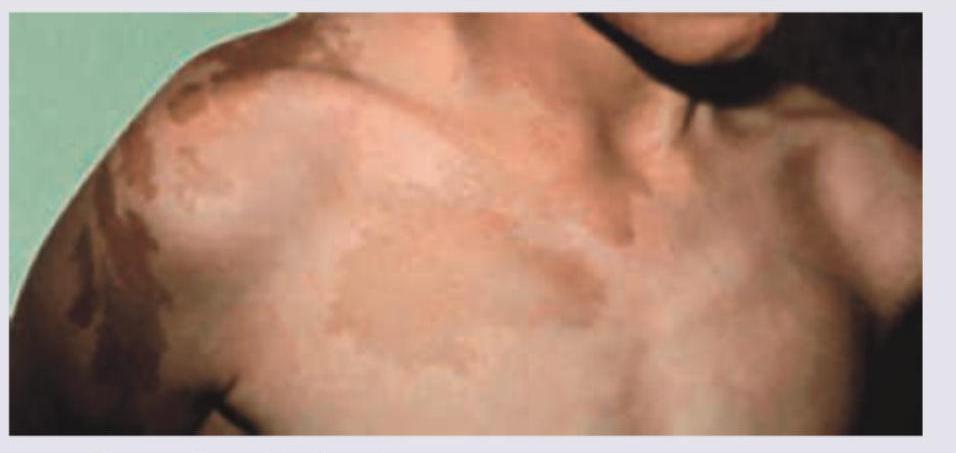
A 5-year-old girl is referred for vaginal bleeding. Physical examination shows breast development, multiple cystic changes in bone radiologically and following skin finding. What is the most likely pubertal disorder?
Practice by Chapter
Disorders of Growth
Practice Questions
Thyroid Disorders
Practice Questions
Disorders of Puberty
Practice Questions
Adrenal Disorders
Practice Questions
Diabetes Mellitus in Children
Practice Questions
Disorders of Calcium and Phosphate Metabolism
Practice Questions
Disorders of Sexual Development
Practice Questions
Hypoglycemia
Practice Questions
Obesity and Metabolic Syndrome
Practice Questions
Pituitary Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Endocrine Emergencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app