
Endocrinology — MCQs

On this page
What is the most common cause of death in children with Diabetic ketoacidosis?
Which of the following is the most common cause of mortality in diabetic ketoacidosis?
Which of the following is not true in Turner Syndrome?
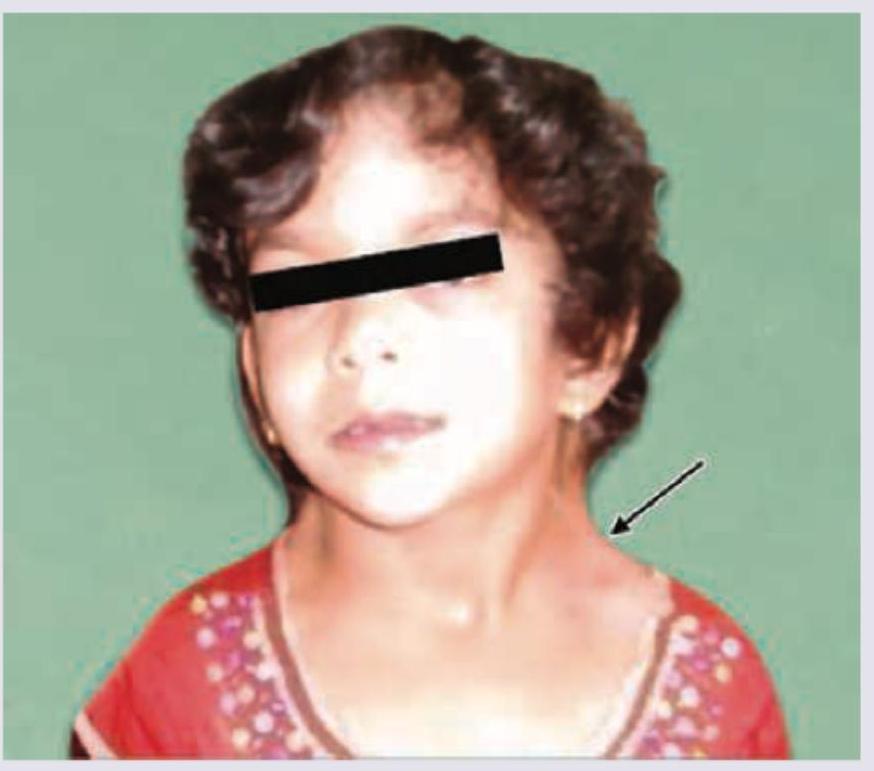
A female baby of 6 years presents with the features shown in image. All the following statements regarding this condition are true except:
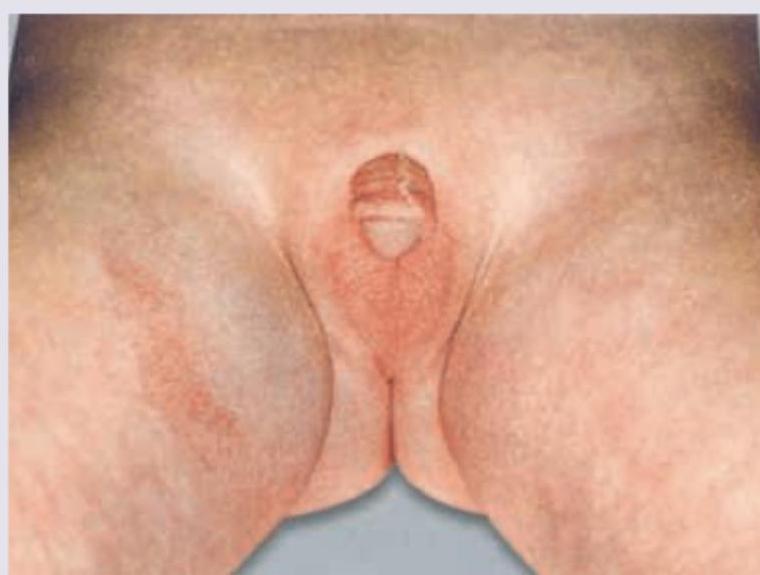
A Lethargic hypoglycemic girl neonate has the following genital appearance. What is the probable cause? (Recent Neet Pattern 2016-17)
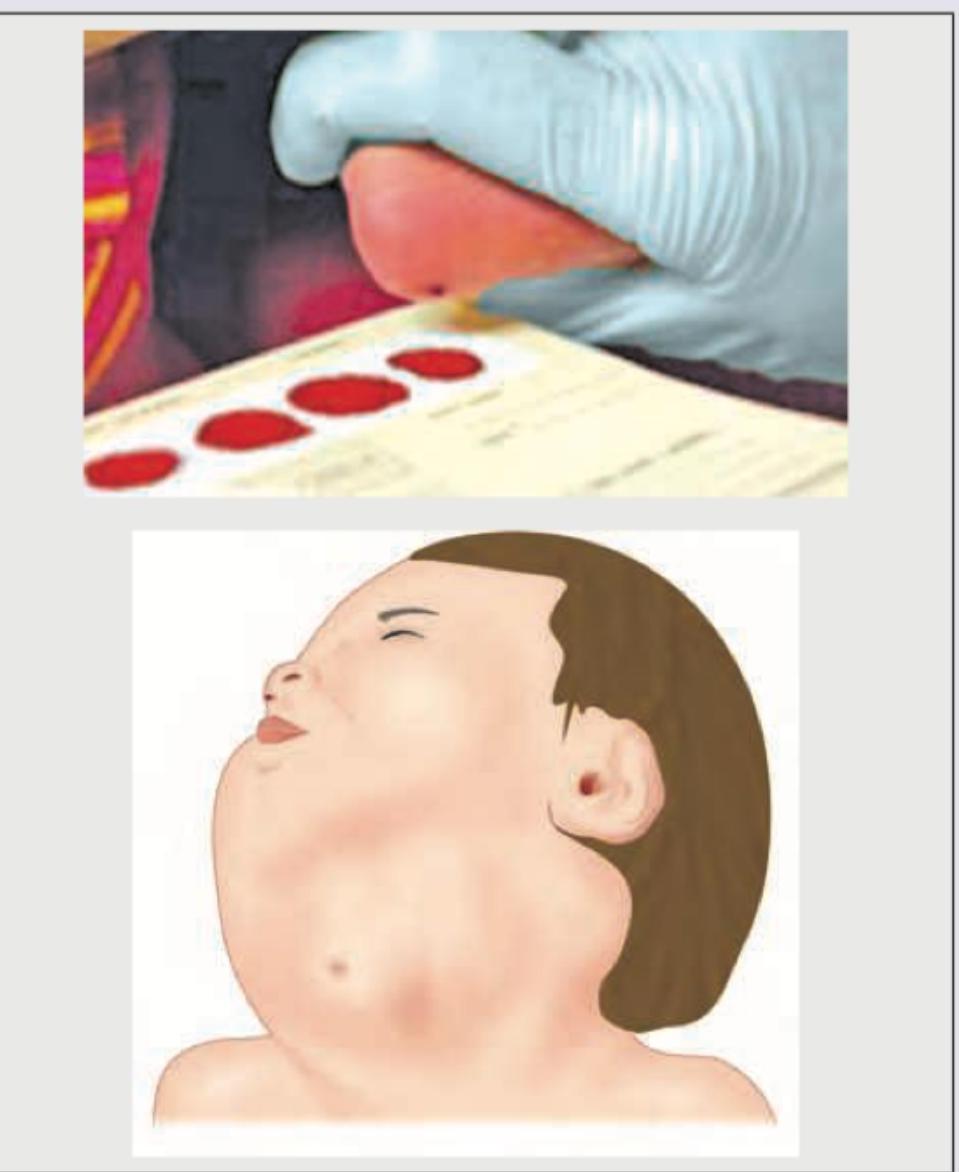
In a neonate with the following findings, what is the likely etiology of hypothyroidism? (NEET Pattern 2018)
A 5-year-old girl presents with vaginal bleeding and thelarche. An X-ray shows multiple cysts in long bones, and she has multiple hyper-pigmented spots. This presentation is suggestive of McCune Albright syndrome. What type of precocious puberty is associated with this syndrome?
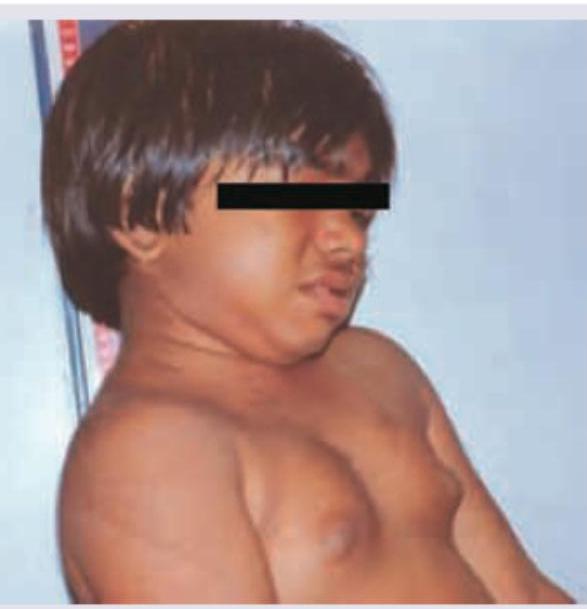
The image shows a child with virilisation and clitoromegaly. What laboratory finding is typical for this condition, assuming the most common enzyme defect?
The following instrument is used for:

All of the following associations are seen with this syndrome except:
Practice by Chapter
Disorders of Growth
Practice Questions
Thyroid Disorders
Practice Questions
Disorders of Puberty
Practice Questions
Adrenal Disorders
Practice Questions
Diabetes Mellitus in Children
Practice Questions
Disorders of Calcium and Phosphate Metabolism
Practice Questions
Disorders of Sexual Development
Practice Questions
Hypoglycemia
Practice Questions
Obesity and Metabolic Syndrome
Practice Questions
Pituitary Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Endocrine Emergencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app