
Molecular Pathology — MCQs

On this page
Edward syndrome is characterized by which of the following chromosomal abnormalities?
All are present in Fragile X syndrome, except:
Mutation in Marfan's syndrome is in which gene?
The following karyotype is suggestive of which disorder?
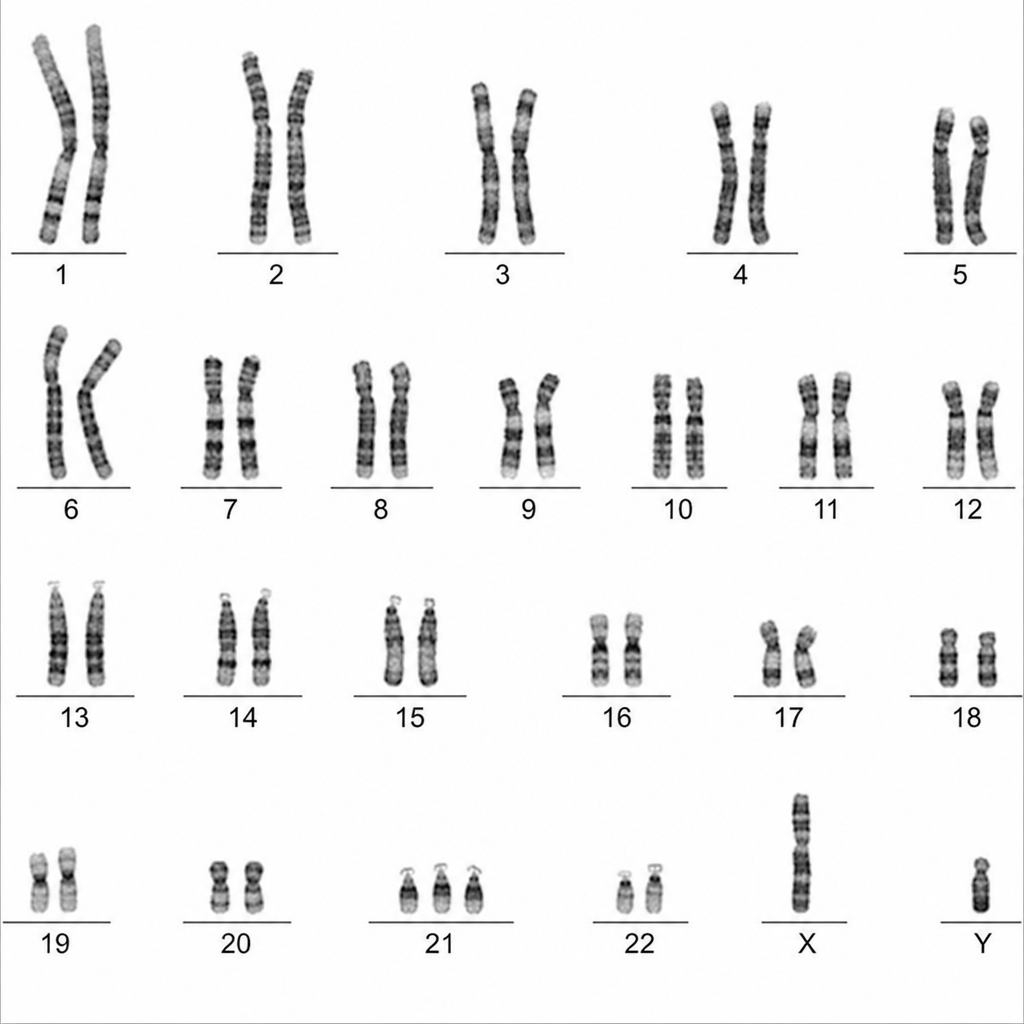
What is the approximate resolution of visualizing chromosomes through a light microscope?
Which of the following is a clinical feature of 22q11 deletion syndrome?
What is the most common trisomy among the following?
Which one of the following statements is FALSE regarding autosomal dominant disorders?
Premutation is seen in which of the following conditions?
Karyotyping is done with all, except:
Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app