
Molecular Pathology — MCQs

On this page
Which of the following is NOT an X-linked disorder?
BRAF V600E mutations are seen in all of the following except?
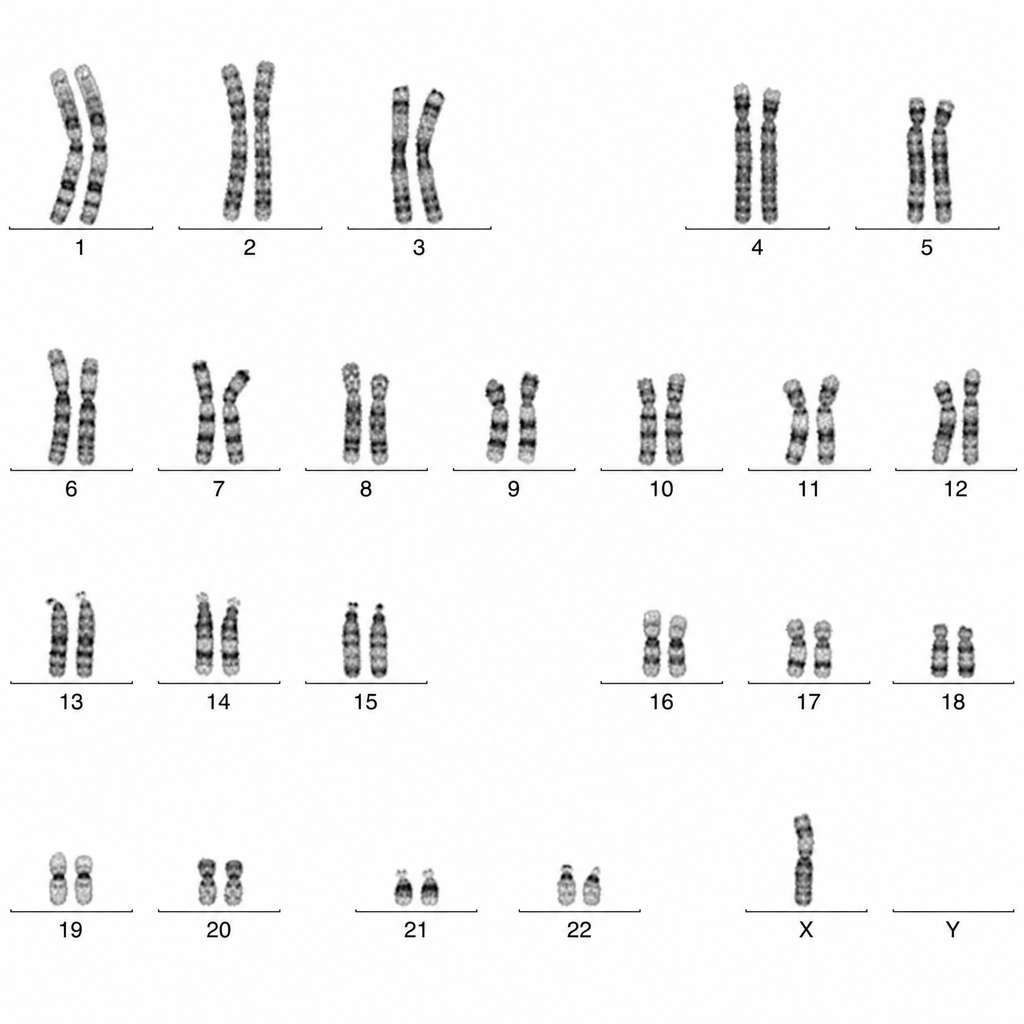
A female patient's karyotype is shown. What is the expected clinical abnormality?
In an autosomal recessive disorder, if one parent is normal and the other is a carrier, and the child is affected, what is the genetic mechanism responsible?
Which of the following is true regarding Turner syndrome?
A couple has two children affected with tuberous sclerosis. On detailed clinical and laboratory evaluation (including molecular studies), both parents are normal. Which one of the following explains the occurrence of two affected children in this family?
Males who are sexually underdeveloped with rudimentary testes and prostate glands, sparse pubic and facial hair, long arms and legs and large hands & feet are likely to have which chromosomal abnormality?
Angelman syndrome is caused by:
Which of the following tests is not used for the detection of specific aneuploidy?
A particular genetic disorder appears in three consecutive generations of a family without any sex predilection. It was also noticed that phenotypically normal family members were having healthy offspring. What is the pattern of inheritance of this disorder?
Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app