
Molecular Pathology — MCQs

On this page
Which of the following conditions CAN BE transmitted as a recessive, sex-linked trait? I. Retinitis pigmentosa II. Colour blindness III. Cystic fibrosis IV. Duchenne muscular dystrophy Select the correct answer using the code given below :
Which of the following syndromes are caused due to genomic imprinting? I. Rubinstein Taybi syndrome II. Prader-Willi syndrome III. Angelman syndrome IV. Edward syndrome Select the correct answer using the code given below :
Which one of the following correctly denotes the inheritance pattern of cystic fibrosis?
Which one of the following is an autosomal recessive disease?
Which one of the following is due to the monosomy of X-chromosome?
A 23-year-old female with a height of 4 feet has a karyotype as shown in the image below. Which among the following indicates the correct etiology?
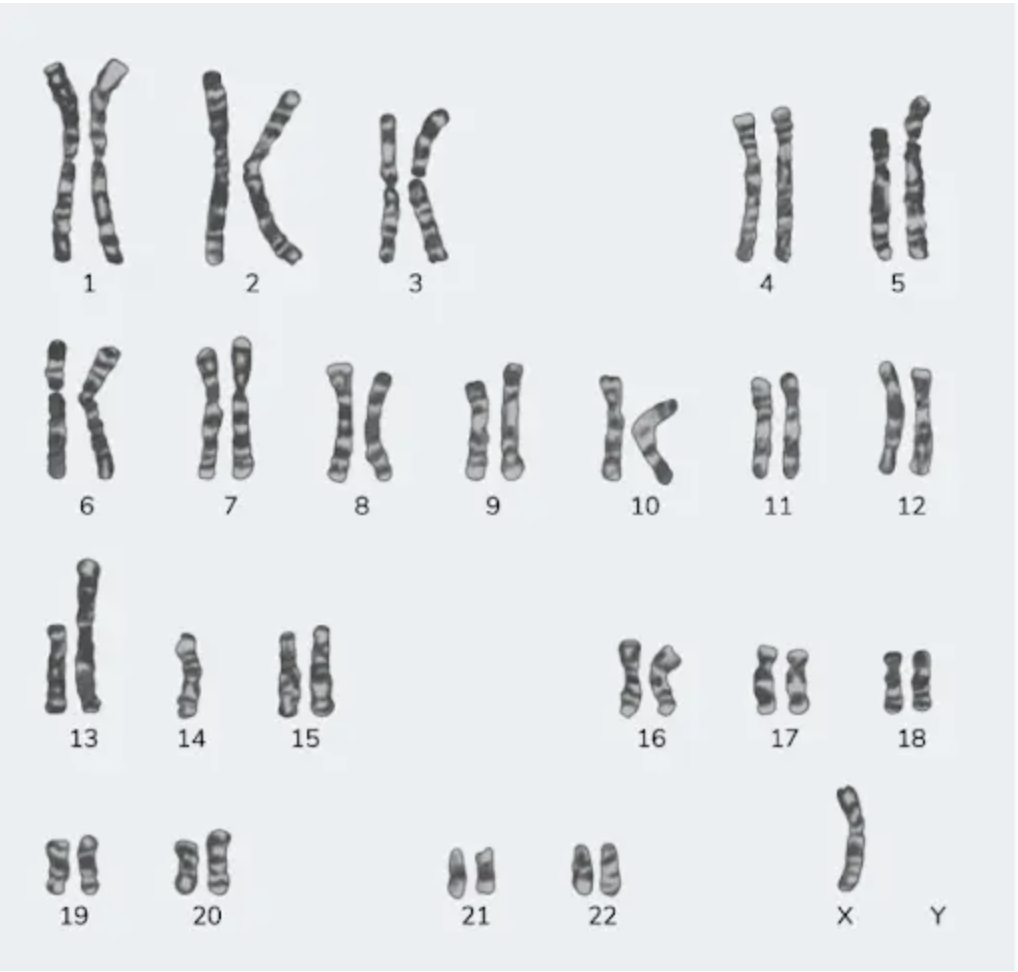
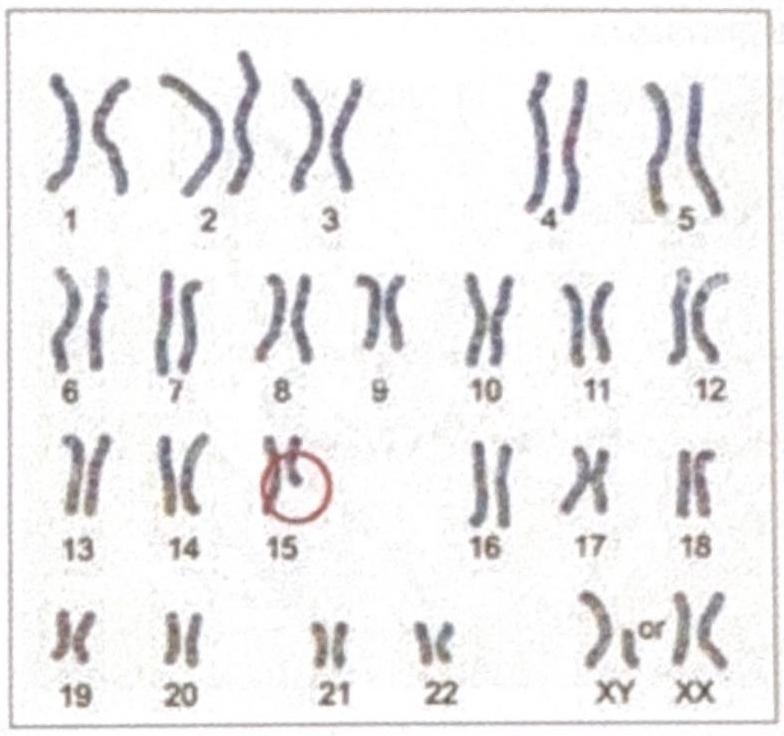
The diagrammatic representation of the karyotype of an individual indicates a specific genetic abnormality. What is the diagnosis?
HNPCC has defect in which
Which of the following is associated with defect in mismatch repair
Best method for the detection of mutations with low allele frequency is:
Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app