
Molecular Pathology — MCQs

On this page
A child presents with a right transverse palmar crease, a cardiac defect, survival into childhood, and mild intellectual impairment. What chromosomal mechanism is most likely associated with these findings?
A child presents with micrognathia and low-set ears. These clinical features are commonly associated with which type of genetic abnormality?
Which gene would you test if the patient has a family history of breast and ovarian cancer?
Which of the following is not an aneuploidy?
A 15-year-old tall boy with long limbs presents to the OPD. On ocular examination, bilateral ectopia lentis is noted. Which gene is most likely affected in this inherited disorder?
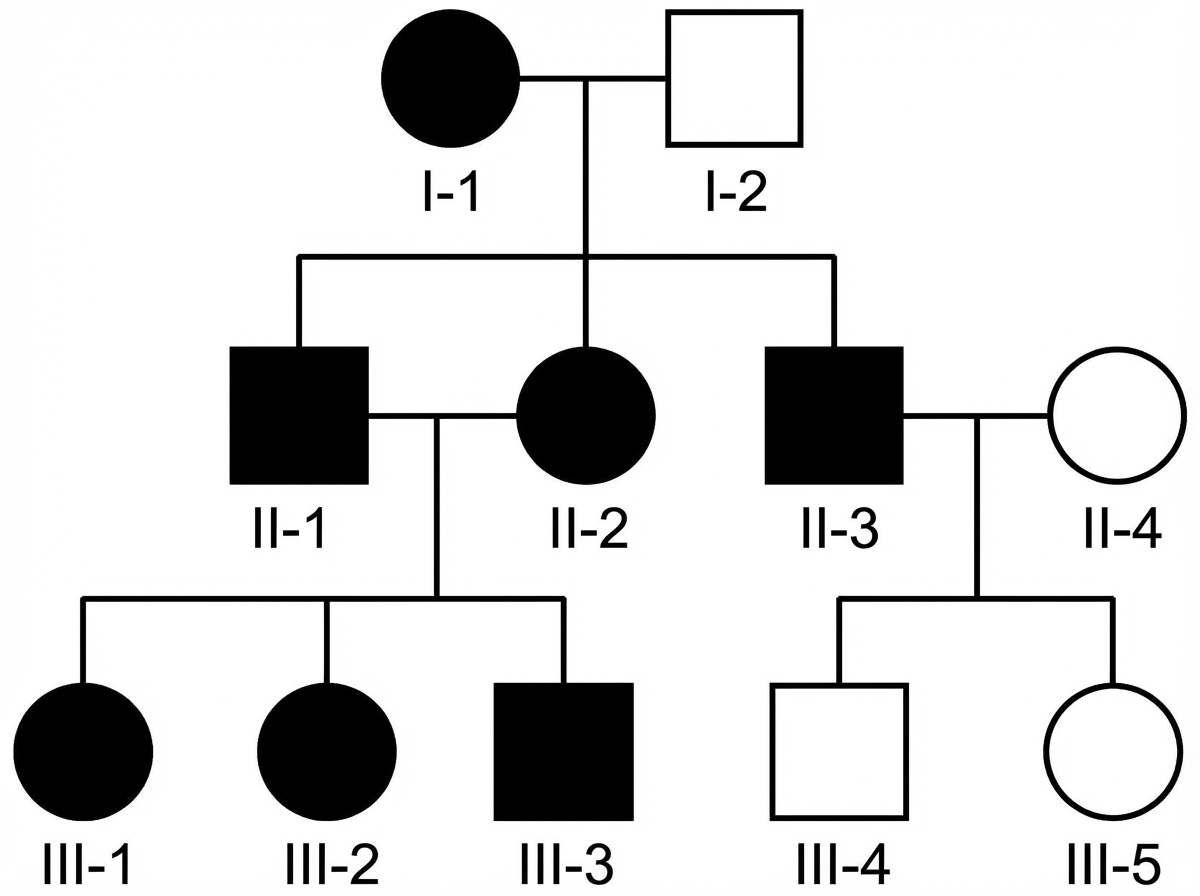
The pedigree chart shown in the image demonstrates a specific pattern of inheritance. Which of the following conditions is most likely to follow this pattern of inheritance?
A 22-year-old tall male presents with long limbs, increased arm span, hypermobile joints, and high-arched palate. On examination, he has lens subluxation and a diastolic murmur suggestive of aortic root dilation. Which of the following genes is most likely mutated in this condition?
Which of the following is an incorrect gene-disease association?
The karyotypic anomaly in the given image denotes which of the following diseases?
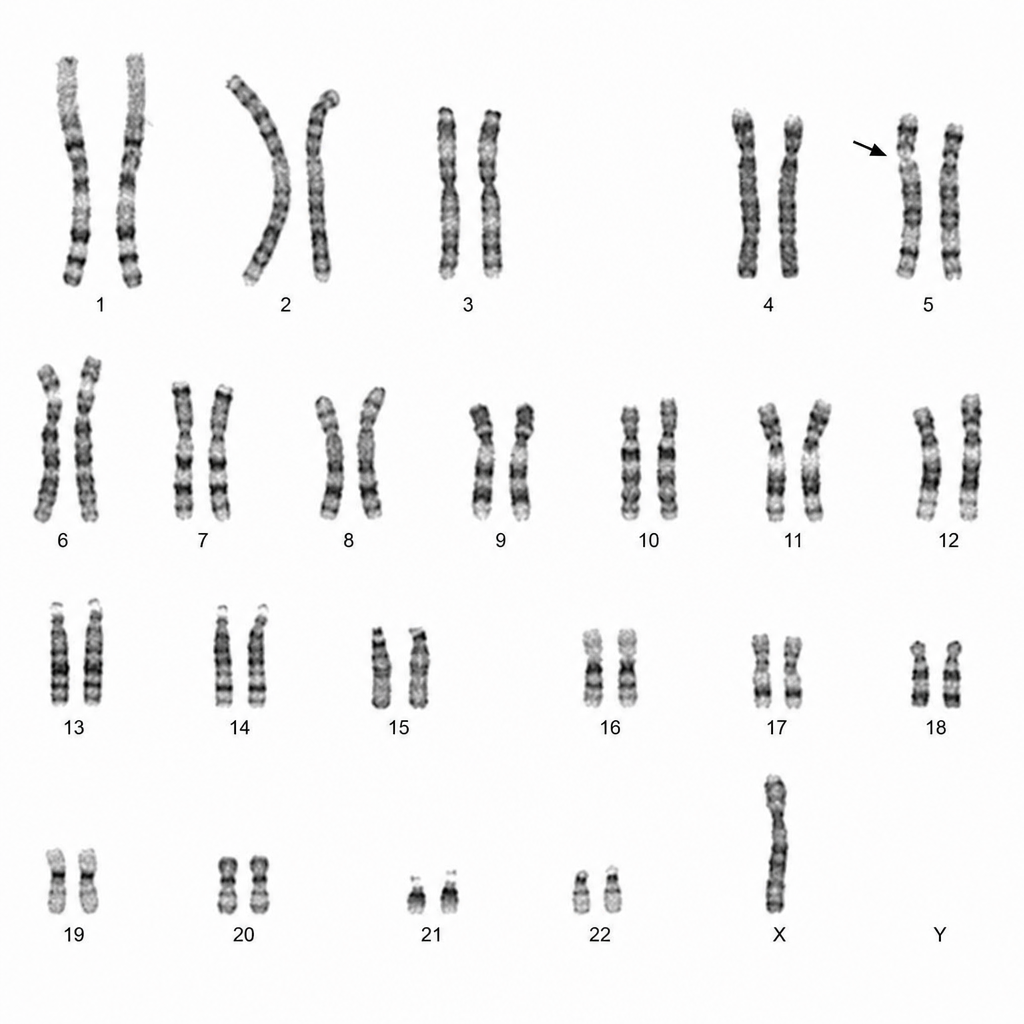
A karyotype is shown in the image. What is the most likely diagnosis?

Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app