
Molecular Pathology — MCQs

On this page
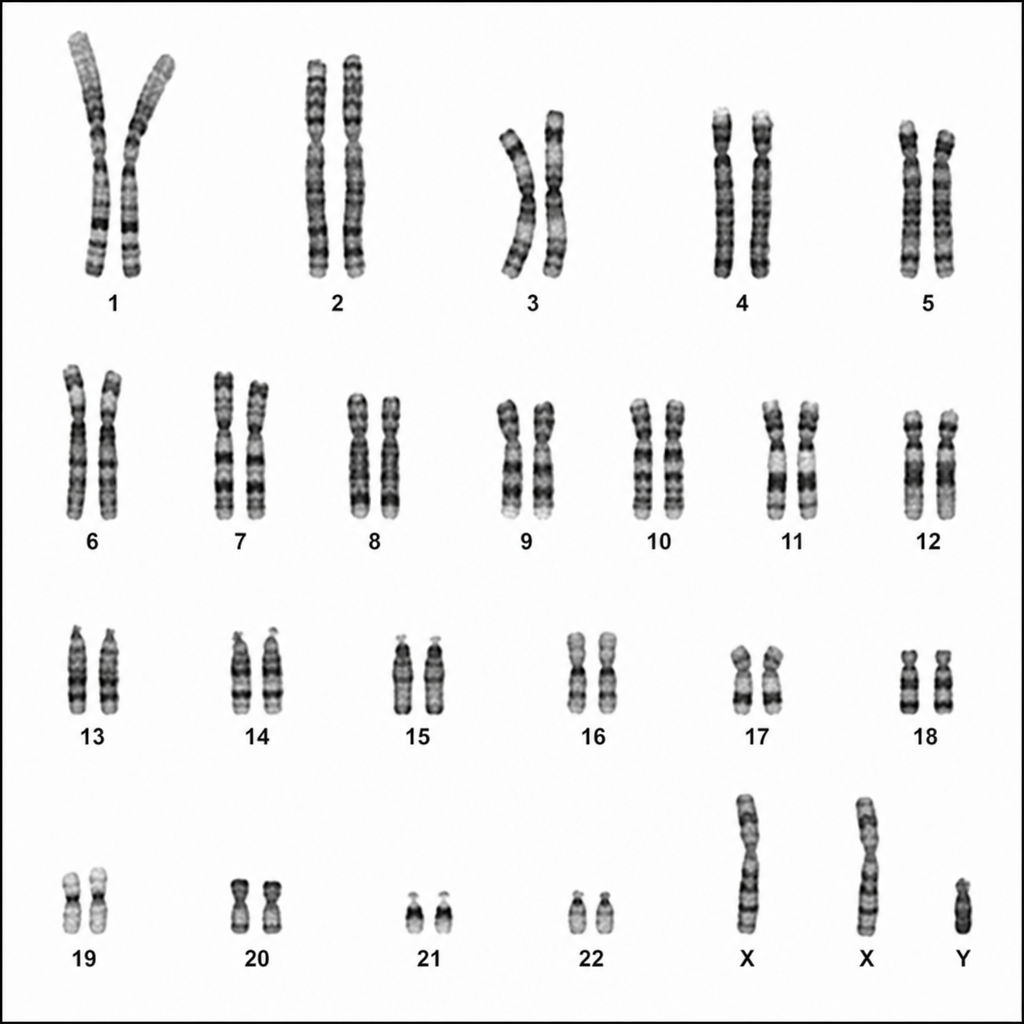
Which of the following chromosomal abnormalities is indicated by the chromosomal mapping?
Hereditary retinoblastomas develop due to an abnormality in which of the following chromosomes?
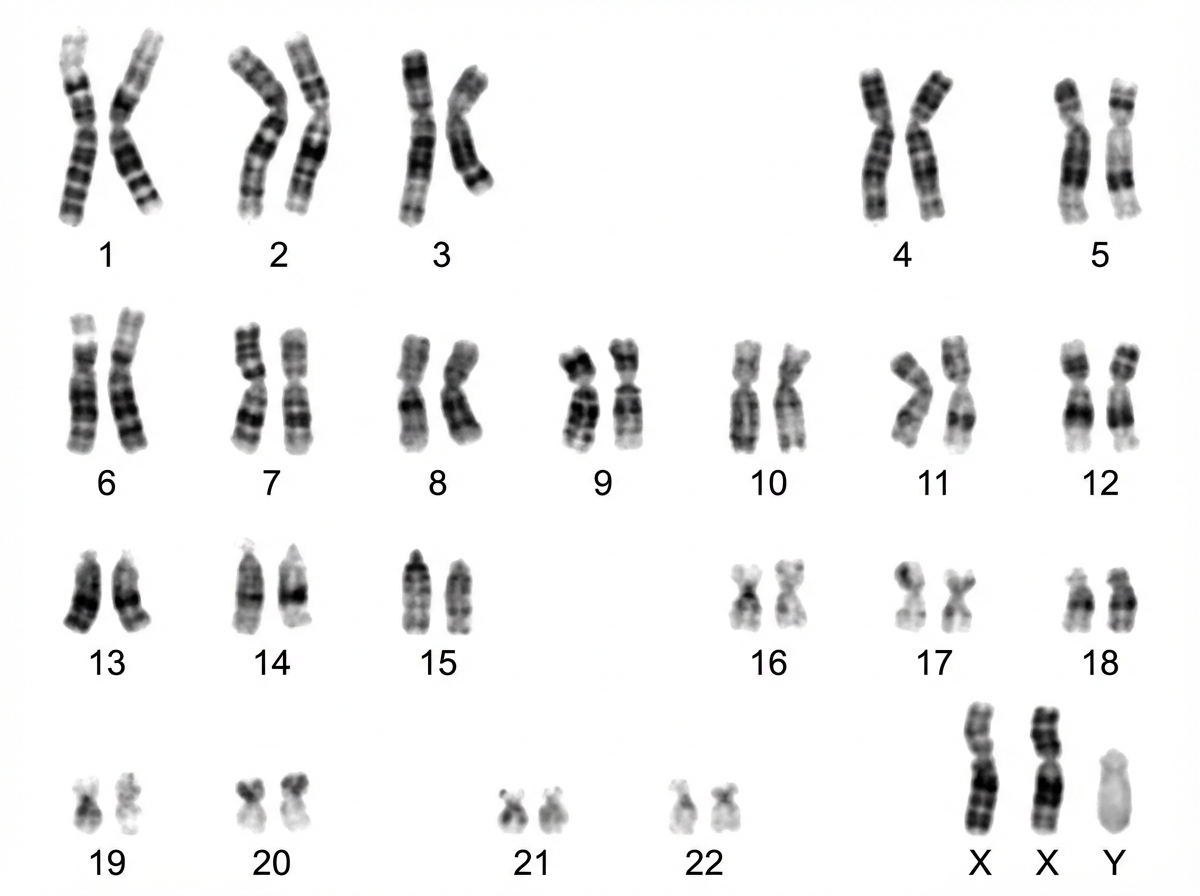
In the provided karyotype, which abnormality is observed? The karyotype shows 2 X-chromosomes and 1 Y-chromosome.
All of the following have autosomal dominant pattern of inheritance except?
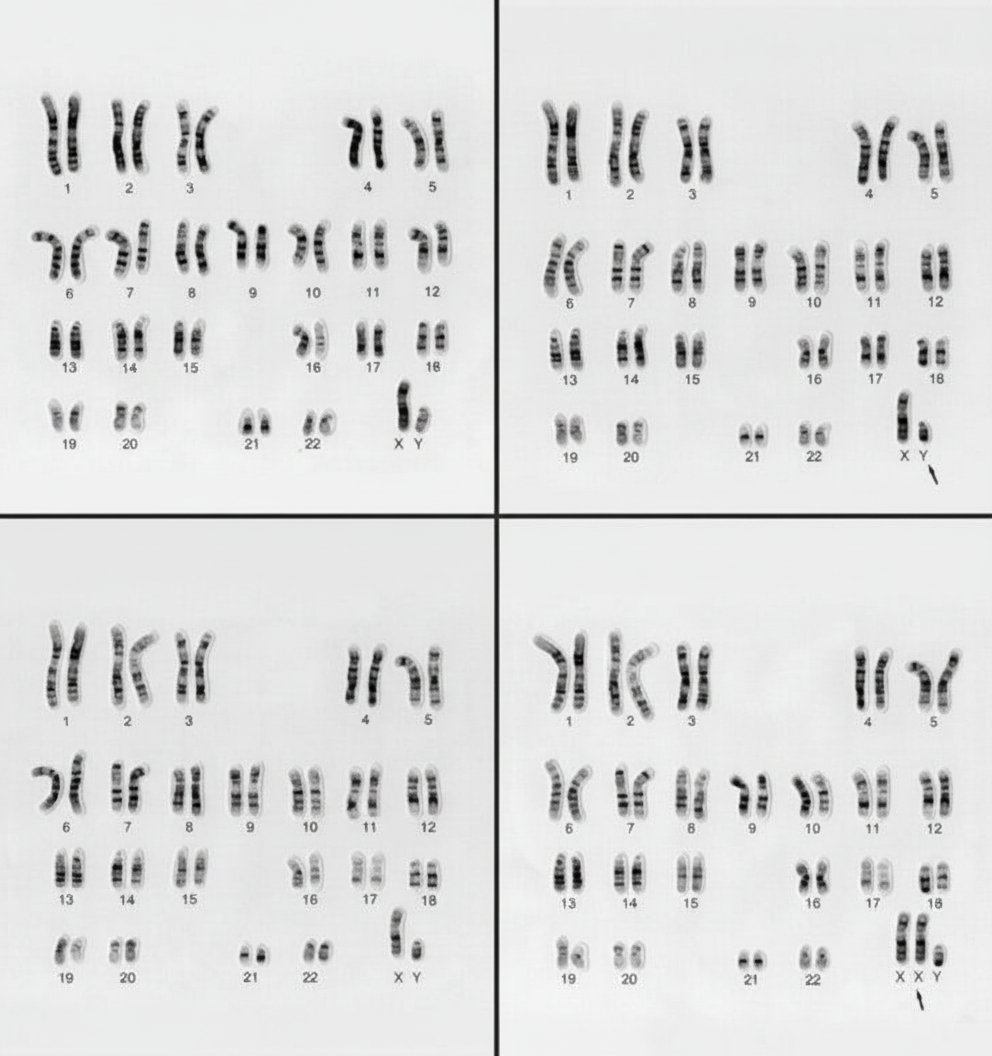
What is the abnormality seen in the following karyotype (lower-right panel)?
AKT1 E17K somatic mutation is associated with which of the following carcinomas?
Which of the following is NOT true about chromosomal instability syndromes?
The NF-2 gene codes for which protein?
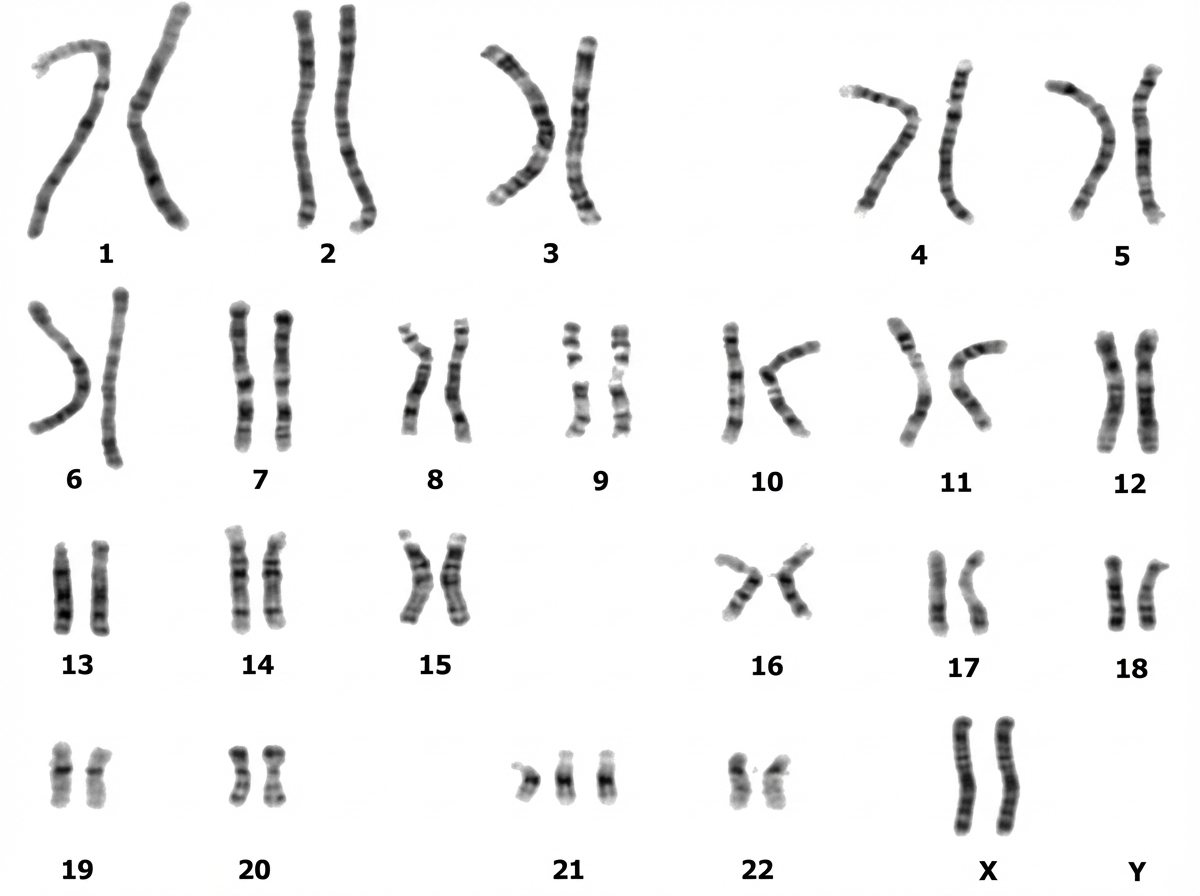
This gene mapping indicates:
Defective DNA repair is seen in which of the following conditions?
Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app