
Molecular Pathology — MCQs

On this page
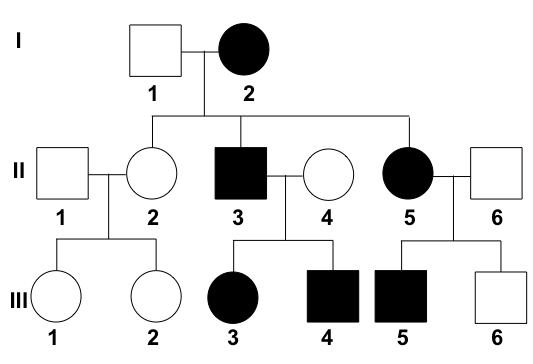
Analyze the provided pedigree chart to identify the underlying genetic disease.
What is the most common tumor in females with tuberous sclerosis?
What is a Leiden mutation?
Aneuploidy is seen with all except:
A 20-year-old woman has a Robertsonian translocation involving chromosome 21 and a second acrocentric chromosome. What is the theoretic likelihood of a functional trisomy 21 if one of her ova is fertilized by a normal sperm?
Trisomy 13 is identified as:
Which gene mutation is seen in Marfan syndrome?
Von Hippel-Lindau disease is associated with all of the following except?
What is the function of the FMR1 protein?
Which of the following statements regarding Down syndrome is FALSE?
Practice by Chapter
Principles of Molecular Pathology
Practice Questions
DNA and RNA Analysis Techniques
Practice Questions
Cytogenetics
Practice Questions
Polymerase Chain Reaction Applications
Practice Questions
Next-Generation Sequencing
Practice Questions
Molecular Diagnosis of Infectious Diseases
Practice Questions
Molecular Oncology
Practice Questions
Pharmacogenomics
Practice Questions
Genetic Counseling and Risk Assessment
Practice Questions
Molecular Diagnostics Quality Control
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app