
Liver and Biliary Pathology — MCQs

On this page
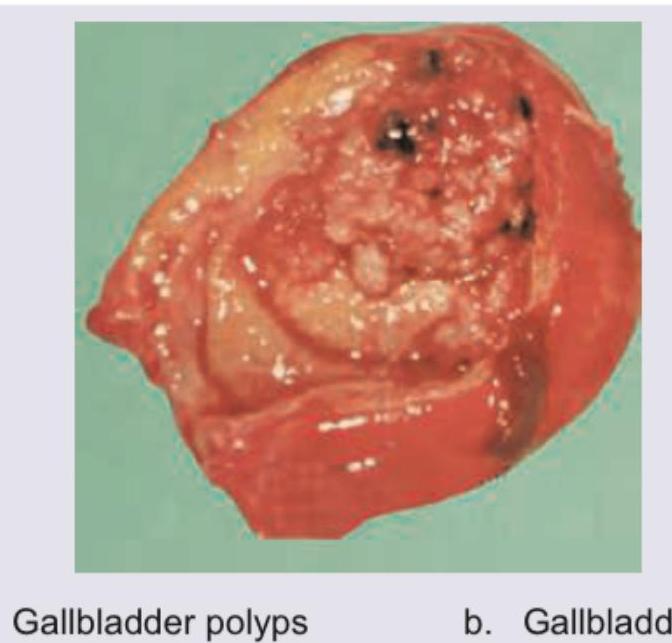
A patient with symptomatic cholecystitis underwent a cholecystectomy. The following specimen was obtained. What is the diagnosis?
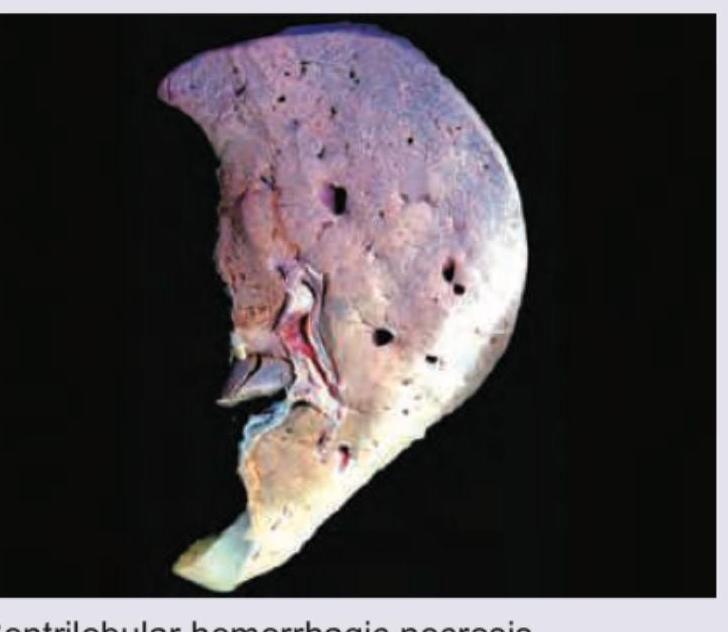
All are true about this liver specimen except:
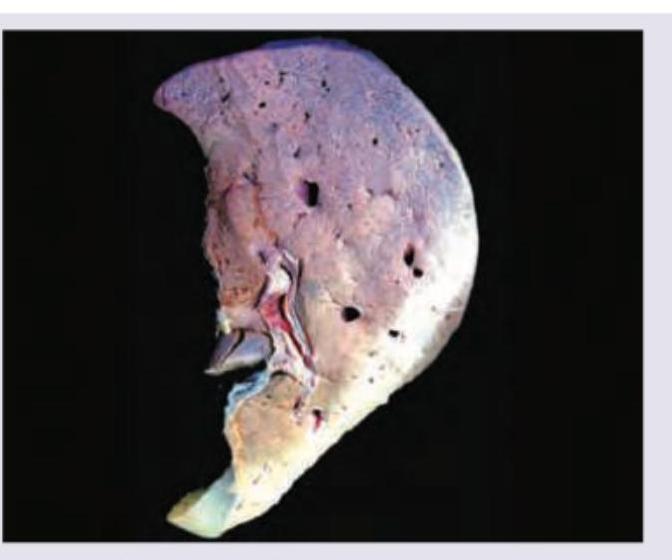
The following liver specimen shows:
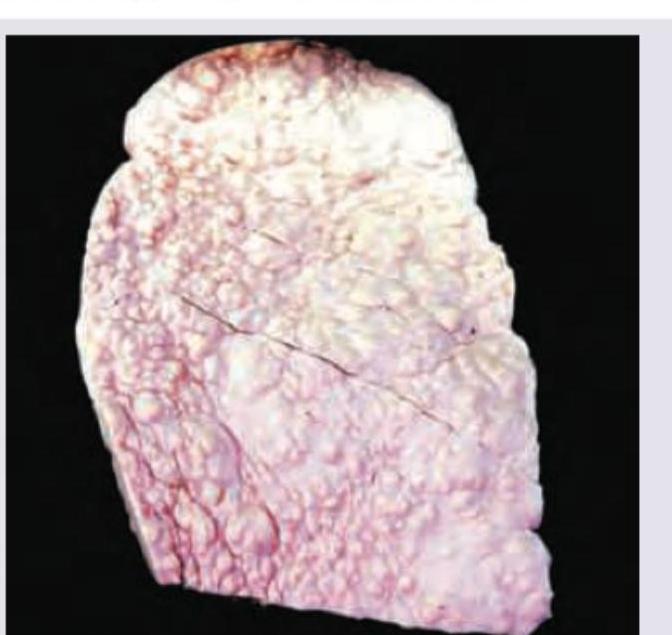
The liver specimen is diagnostic of:
All are true about the marking X in histopathological specimen from a patient of fatty liver except:
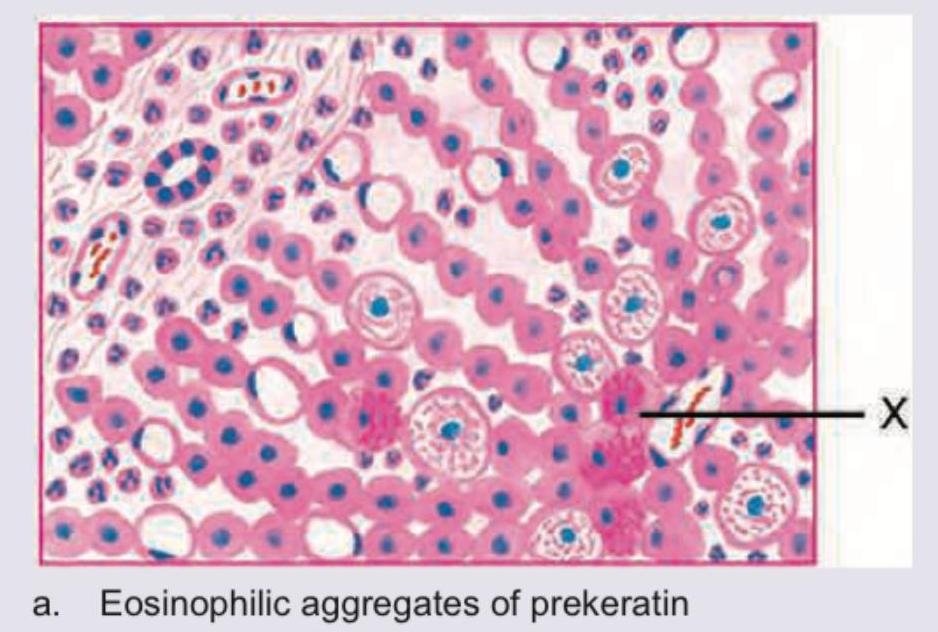
The marking X in a histopathological specimen from a patient of fatty liver shows:
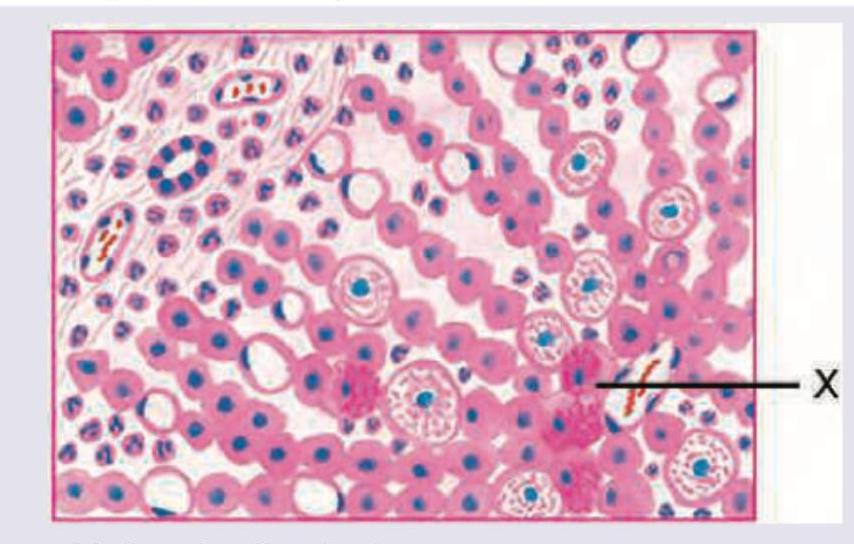
A 30-year-old shepherd presents with painless hepatomegaly. Peripheral blood work shows eosinophilia. The histopathological slide of surgically removed cyst shows a marking X. This denotes:
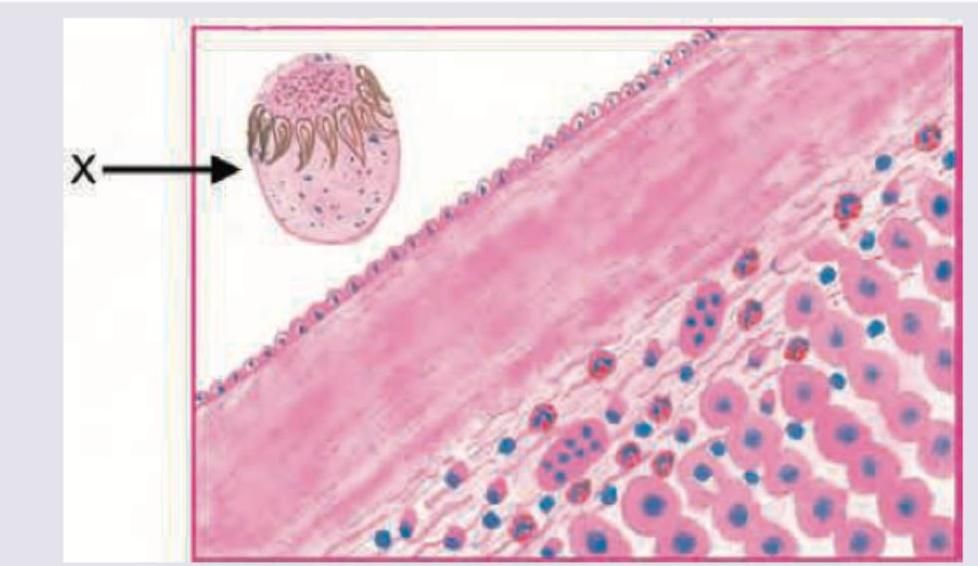
A 20-year-old college student presents with 7 day history of nausea and feeling feverish. He has tenderness in right upper quadrant and admits to high risk sexual behavior. The liver biopsy marking $X$ shows:
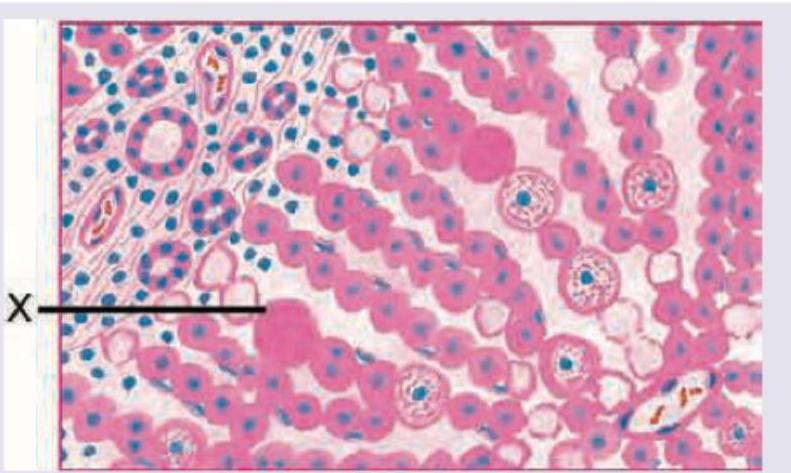
A 54-year-old chronic alcoholic died due to a liver disease. His pathological specimen is provided in the image below. The most likely diagnosis is:

A 14-year-old male child presents with right upper quadrant pain, his pathological presentation is given in the image. Which one of the following statements is false?

Practice by Chapter
Jaundice and Cholestasis
Practice Questions
Viral Hepatitis
Practice Questions
Alcoholic and Non-alcoholic Fatty Liver Disease
Practice Questions
Drug and Toxin Induced Liver Injury
Practice Questions
Cirrhosis and Its Complications
Practice Questions
Metabolic Liver Diseases
Practice Questions
Liver Tumors
Practice Questions
Gallbladder and Biliary Tract Diseases
Practice Questions
Congenital Liver Diseases
Practice Questions
Liver Transplantation Pathology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app