
Inflammation and Repair — MCQs

On this page
Which of the following statements regarding wound healing is correct?
A patient with long-standing chronic inflammatory disease develops systemic amyloidosis. Which amyloid protein subtype is characteristically deposited?
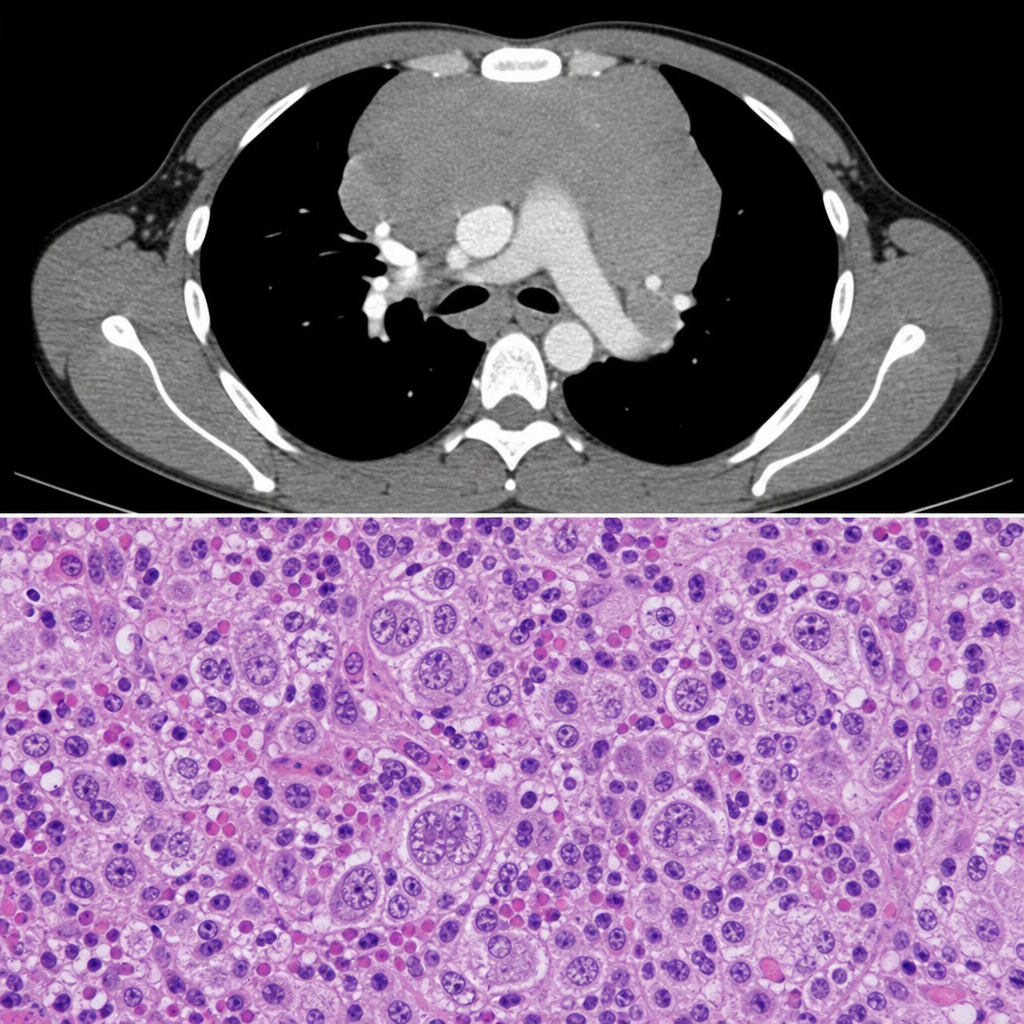
A 22-year-old male presents with painless cervical lymphadenopathy, drenching night sweats, and a 6 kg weight loss over 3 months. CT scan shows mediastinal widening. Excisional lymph node biopsy is sent for histopathology. The biopsy image is shown (Image 2). Immunohistochemistry on the neoplastic cells would most likely demonstrate which of the following marker profiles?
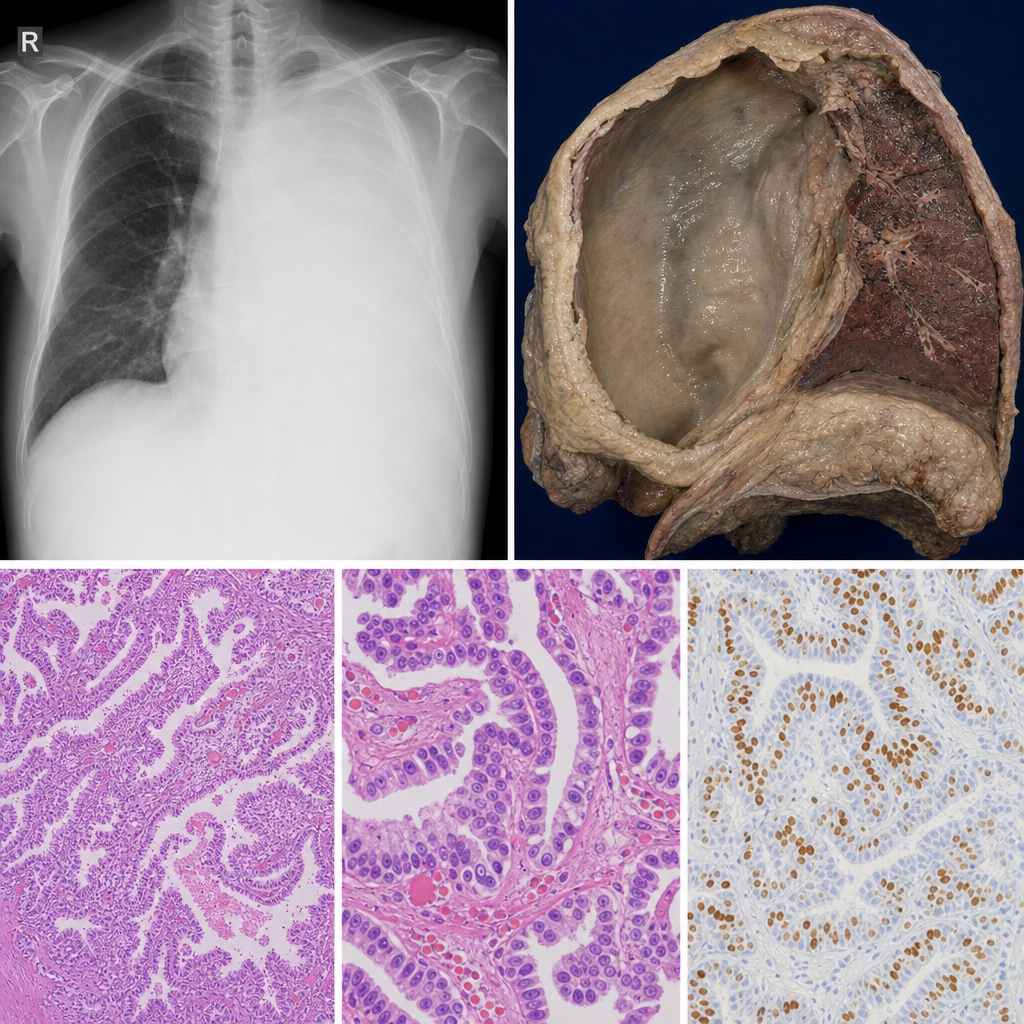
A 62-year-old male shipyard worker presents with progressive dyspnoea and dull chest pain for 6 months. Chest X-ray shows massive unilateral pleural effusion with pleural thickening. He has a 30-year history of occupational dust exposure. The gross pathology specimen is shown in Image 2. Which histological subtype of this tumour carries the best prognosis and is most likely to show strong immunoreactivity for calretinin and WT-1?
Foci of granulomatous inflammation show all of the following except?
Chediak-Higashi syndrome is due to a defect in which of the following processes?
Maximum tensile strength is recovered in a wound area during what period after an injury?
Epithelial granuloma is caused by which of the following immune cells?
Which of the following is considered a major pyrogenic cytokine?
In cellular events of acute inflammation, all of the following are observed EXCEPT:
Practice by Chapter
Acute Inflammation: Vascular Events
Practice Questions
Acute Inflammation: Cellular Events
Practice Questions
Chemical Mediators of Inflammation
Practice Questions
Chronic Inflammation
Practice Questions
Granulomatous Inflammation
Practice Questions
Systemic Effects of Inflammation
Practice Questions
Wound Healing
Practice Questions
Tissue Regeneration
Practice Questions
Fibrosis and Repair
Practice Questions
Resolution of Inflammation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app