
Immunopathology — MCQs

On this page
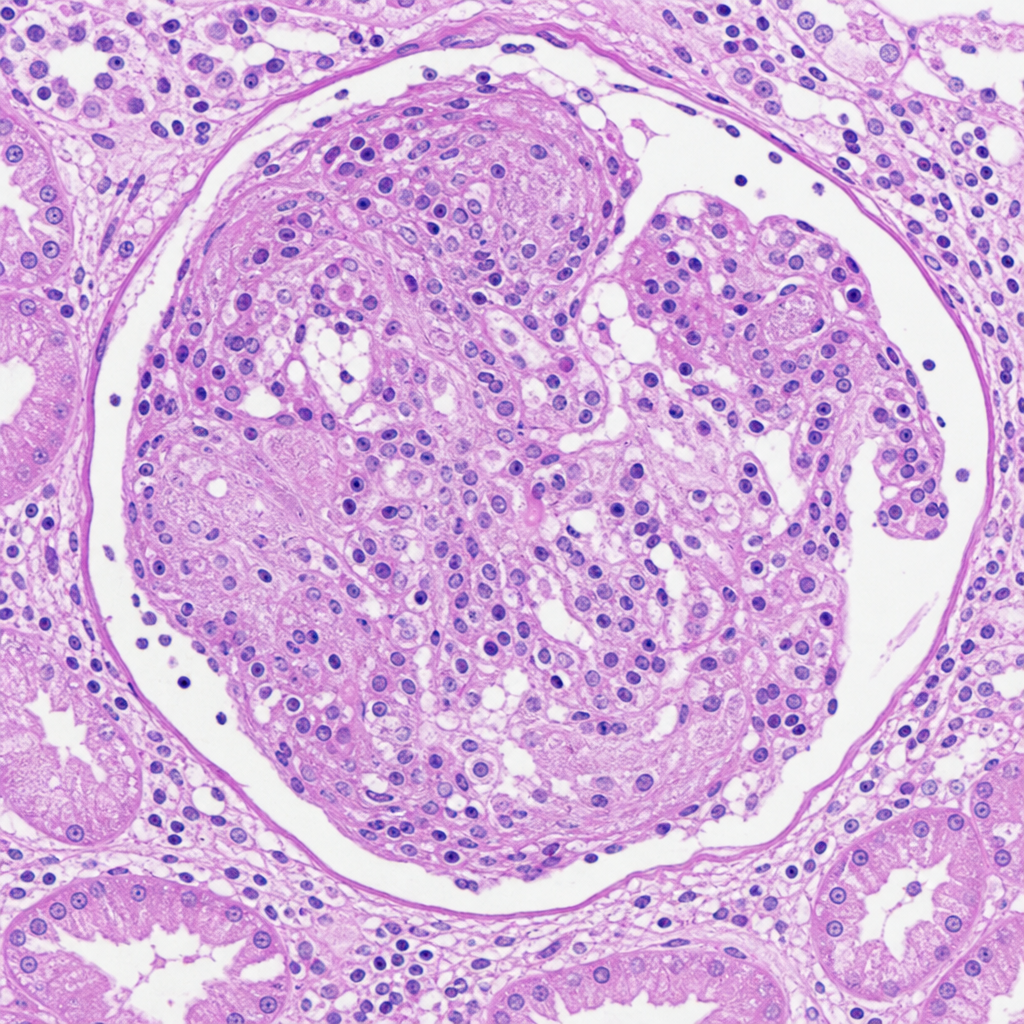
A 30-year-old male presents with rapidly progressive renal failure over 3 weeks, haematuria, and red cell casts in urine. Serum creatinine has risen from 1.2 to 7.8 mg/dL within one month. ANCA is positive. The renal biopsy is shown in Image 3. Which of the following statements best describes the cellular composition and origin of the lesion most prominently seen in this biopsy?
Secondary amyloidosis complicates which of the following conditions?
Hyperacute rejection is due to which of the following mechanisms?
Which of the following cells do not act as antigen-presenting cells?
Which of the following is NOT a disorder of phagocytosis?
Molecular mimicry is an explanation for which of the following?
Erythroblastosis fetalis is an example of which type of hypersensitivity reaction?
Which human leukocyte antigen (HLA) complex is associated with narcolepsy?
A patient on treatment with penicillin developed pallor, but without shortness of breath, urticaria, or wheezing. Investigations revealed antibodies against penicillin in his blood. What is the most likely type of hypersensitivity reaction that occurred in this patient?
Which one of the following are the most important antigen-presenting cells in humans?
Practice by Chapter
Cells and Tissues of the Immune System
Practice Questions
Innate Immunity
Practice Questions
Adaptive Immunity
Practice Questions
Hypersensitivity Reactions
Practice Questions
Autoimmune Diseases
Practice Questions
Immunodeficiency Disorders
Practice Questions
Transplantation Immunopathology
Practice Questions
Immune Response to Infections
Practice Questions
Immunologic Laboratory Techniques
Practice Questions
Tumor Immunology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app