
Hematopathology — MCQs

On this page
Absolute lymphocytosis is seen in which of the following conditions?
A female with recurrent abortion and isolated prolonged APTT is most likely associated with which of the following?
Aplastic anemia is seen in all of the following conditions EXCEPT?
Haptoglobin levels are decreased in which of the following conditions?
Which immunohistochemical marker is most useful in differentiating thymoma from acute lymphoblastic leukemia (ALL)?
A 6-year-old Afroamerican boy presented with abdominal pain, chronic hemolysis, and abnormal RBC shape on peripheral smear. What is the most likely underlying molecular disorder responsible for this condition?
Which of the following is false regarding Transfusion-Related Acute Lung Injury (TRALI)?
Which of the following is not a B cell lymphoma?
Down syndrome predisposes to which type of cancer?
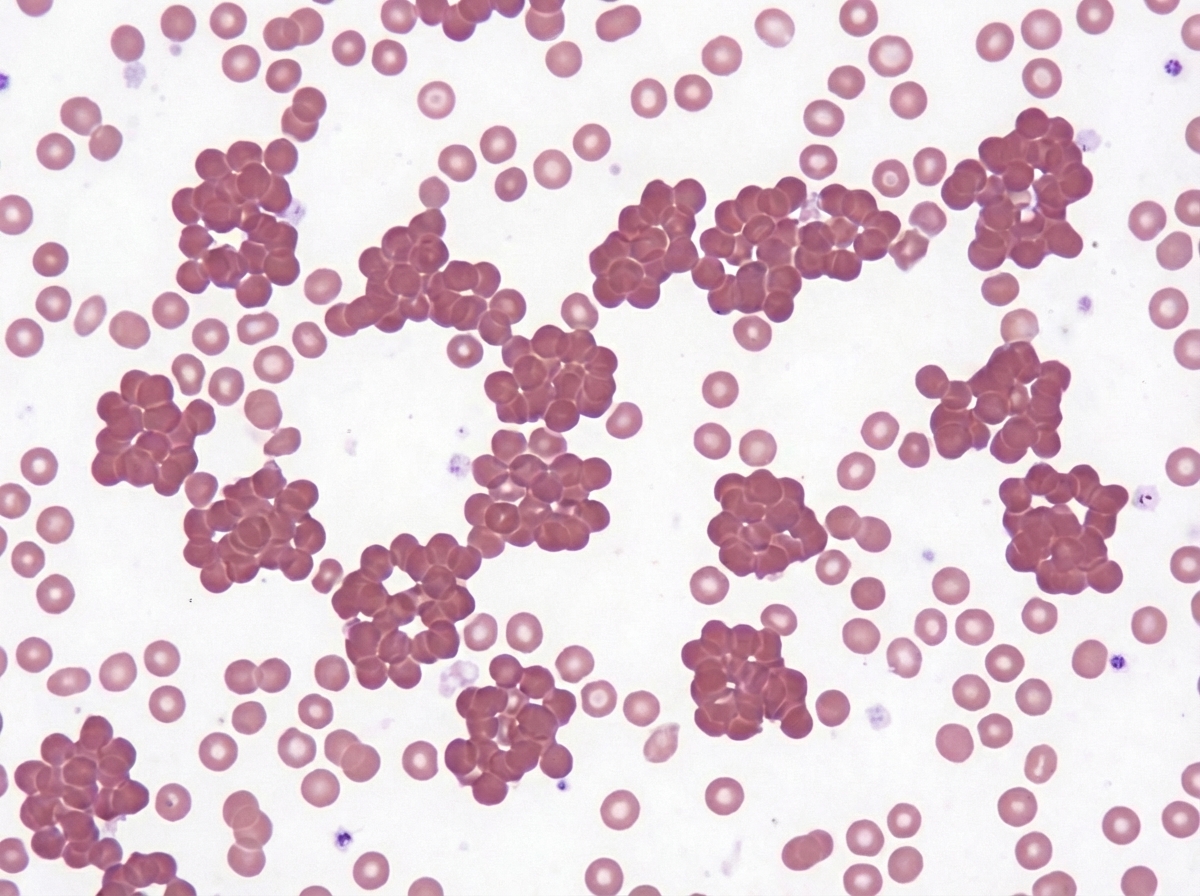
The peripheral blood smear shown in the image is most suggestive of which of the following conditions?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app