
Hematopathology — MCQs

On this page
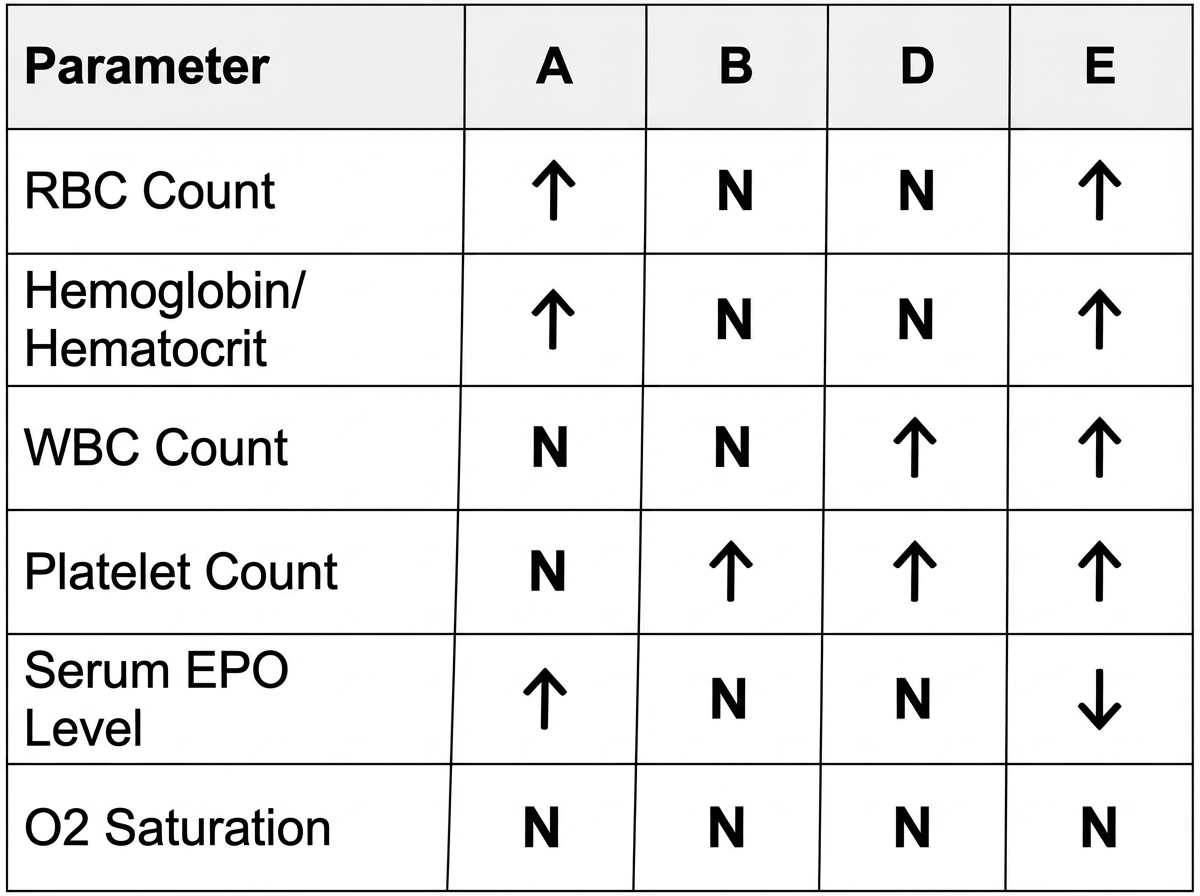
Which one of the labeled boxes in the diagram below is most consistent with the expected findings for an individual with polycythemia rubra vera?
What is the most common extranodal site for non-Hodgkin's lymphoma?
Nuclear-cytoplasmic asynchrony is usually seen in:
What is the most suitable test to assess iron stores?
Acquired mutations in the PIGA gene give rise to which condition?
All of the following are true about nodular sclerosis of Hodgkin's disease except?
Which of the following statements about sickle cell disease is false?
Erythrocyte Sedimentation Rate is zero in which of the following conditions?
What is true about acquired disorders of coagulation?
A 19-year-old man has had recurrent bleeding occur in his knee when playing contact sports. He has no history of spontaneous bleeding, but his brother had similar problems. Consultation with a specialist reveals that he has "mild" hemophilia A. Which of the following factor abnormalities is consistent with this diagnosis?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app