
Hematopathology — MCQs

On this page
All of the following are true regarding the findings of peripheral smear in vitamin B12 deficiency except?
Which of the following lymph nodes are commonly affected in non-Hodgkin's Lymphoma, except?
The peripheral blood smear is characteristic of which of the following hematological conditions?
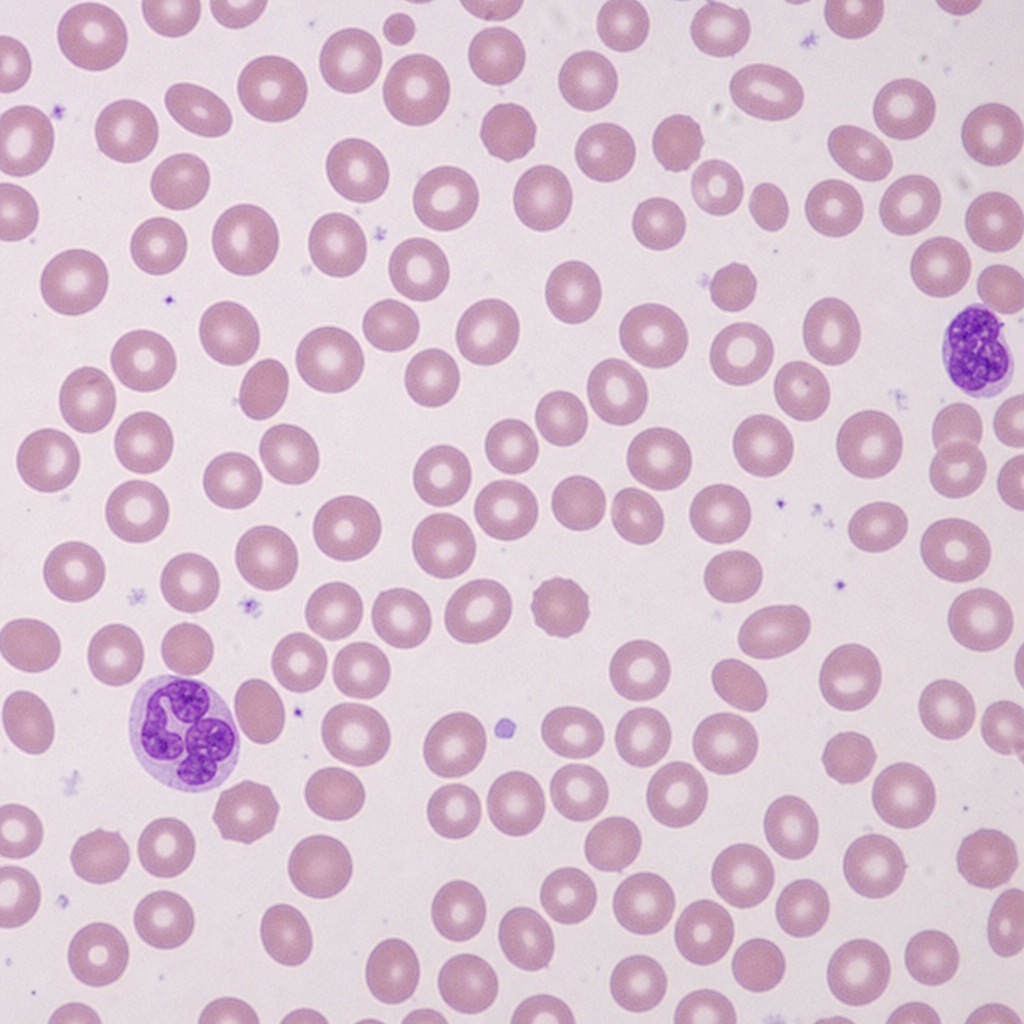
What is the concentration of Hb A2 in thalassemia trait?
CAR-T cells are used in the treatment of which of the following cancers?
Poorest prognosis in Acute Myeloid Leukemia (AML) is seen in which cytogenetic abnormality?
What is the characteristic CD marker for Langerhans cells?
Histopathological examination reveals the diagnosis as:
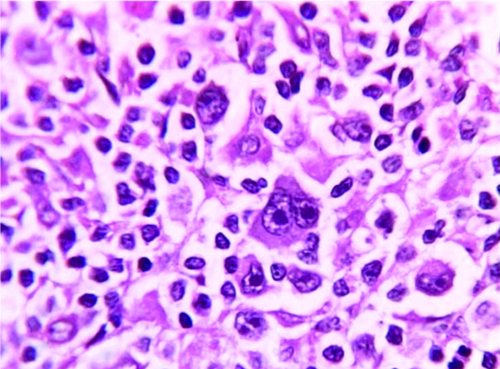
A 65-year-old patient presented with weakness, fatigue for 6 months, and mild abdominal discomfort. Examination revealed moderate splenomegaly. Lab findings showed severe normocytic normochromic anemia, neutropenia with monocytopenia, and thrombocytopenia. Bone marrow aspiration resulted in a 'dry' tap. A bone marrow biopsy was performed. Which of the following CD markers can be positive in this condition EXCEPT?
What is the most common subtype of acute lymphoblastic leukemia (ALL) in children?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app