
Hematopathology — MCQs

On this page
Which of the following is involved in hereditary spherocytosis?
Which of the following is an example of a chronic myeloproliferative disorder?
Routine Rh typing includes testing for which antigen?
A 25-year-old man involved in a motorcycle accident incurs a laceration to his thigh. The bleeding is stabilized en route to the hospital, but on arrival, he is noted to have orthostatic hypotension and his hematocrit is 21%. He receives 2 units of PRBCs. As the first unit is nearly finished transfusing, he becomes febrile and hypotensive. Urine output ceases. The serum above the clot in a red top phlebotomy tube is pink. Which of the following complications of transfusion has most likely occurred in this man?
Hematopoietic stem cells differ from progenitor stem cells in that they can:
What is the most common site of histiocytosis?
Which of the following is false regarding Chediak-Higashi syndrome?
What is the expected reticulocyte count in hemolytic jaundice?
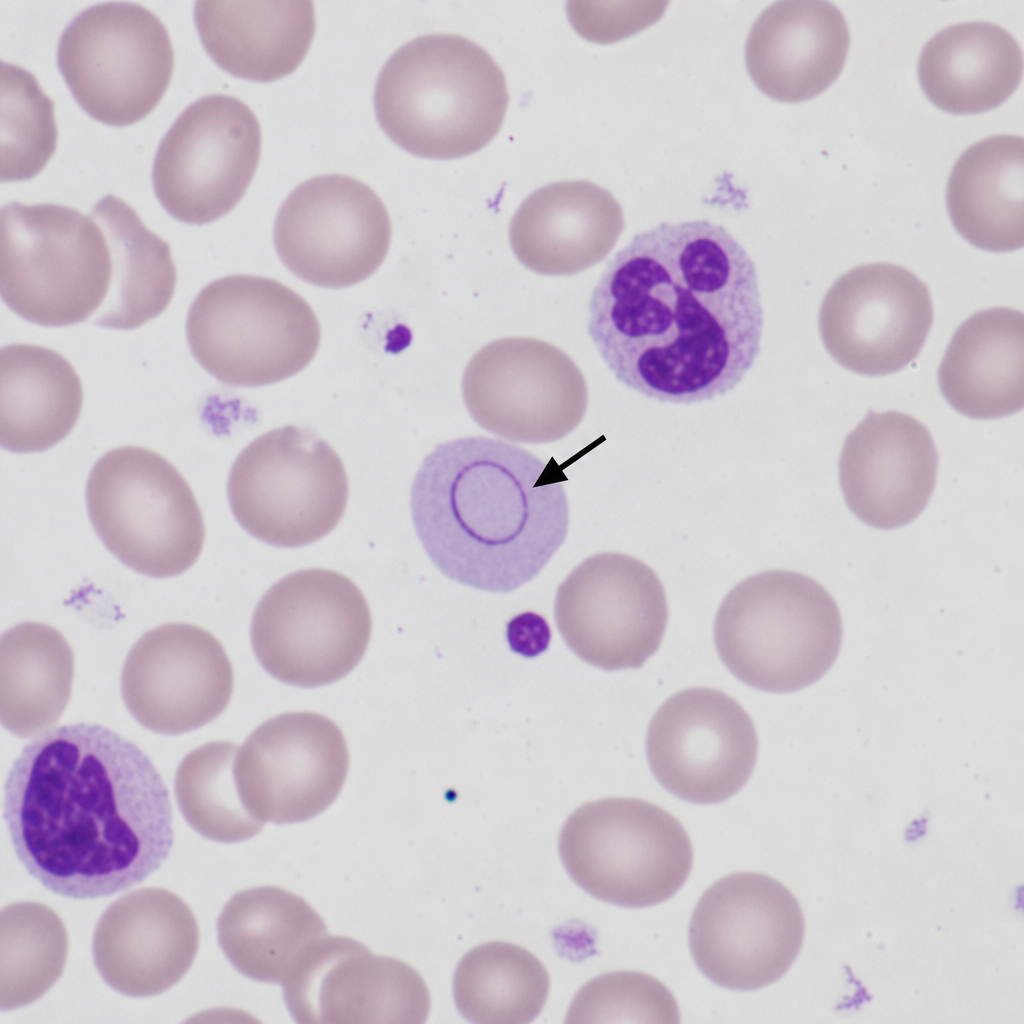
The arrow shows:
Which is the most common antigen implicated in hemolytic disease of the newborn?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app