
Hematopathology — MCQs

On this page
Regarding G6PD deficiency, which of the following is true?
ALL L3 cells resemble which of the following?
What is a common complication seen in polycythemia vera?
Which of the following is the binding site for the von Willebrand factor multimers on the platelets?
A patient presents with microcytic hypochromic anemia. Serum iron levels and TIBC are decreased. What is the likely diagnosis?
A 45-year-old male presents with bony pain, with no history of trauma or drug abuse. Investigations revealed an ESR of 140 mm/hr and Hb of 6 gm/dL. A peripheral smear is shown below. What type of chromosomal abnormality is associated with a favorable prognosis in this patient?
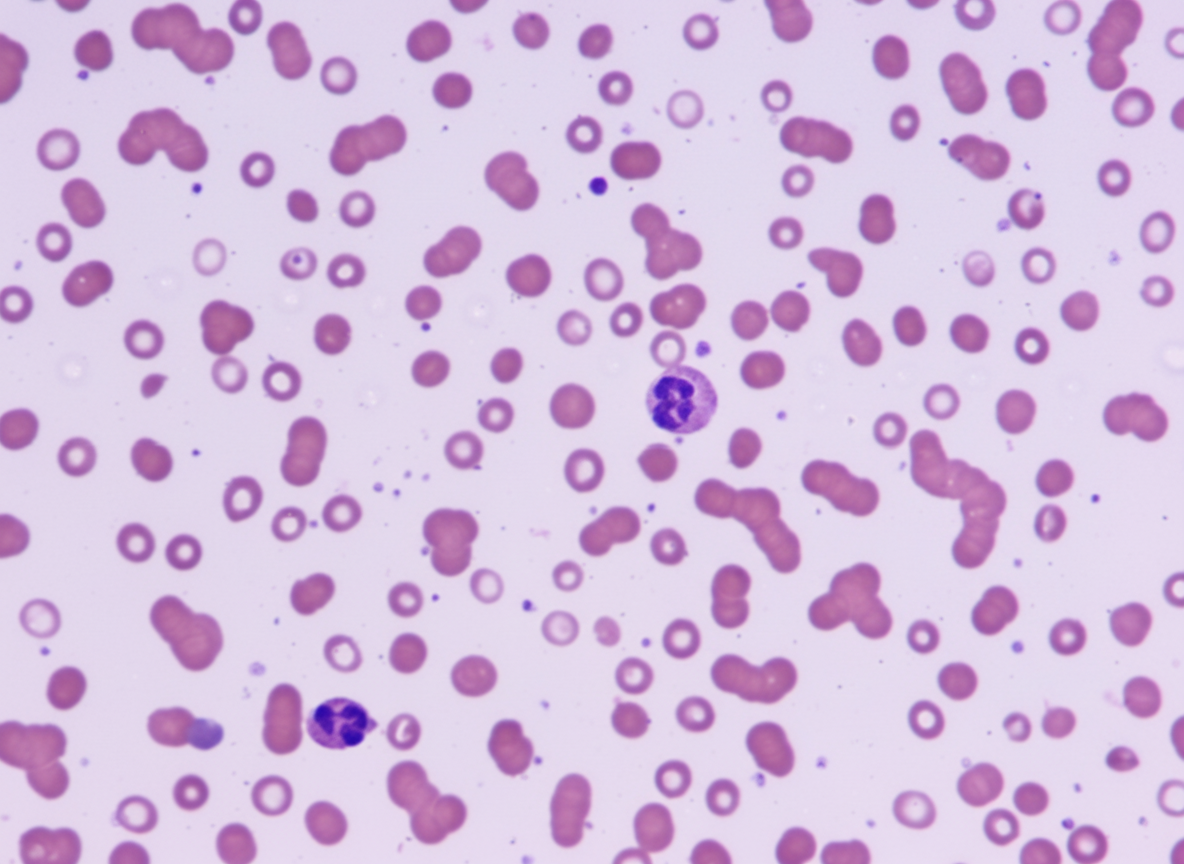
Which of the following disorders has the potential to develop myeloid leukemia?
Lymphoma is caused by all of the following viruses except?
Microcytosis is seen in which of the following conditions?
Which of the following is NOT a characteristic feature of non-Hodgkin lymphoma?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app