
Hematopathology — MCQs

On this page
All of the following are features of hemolytic anemia except?
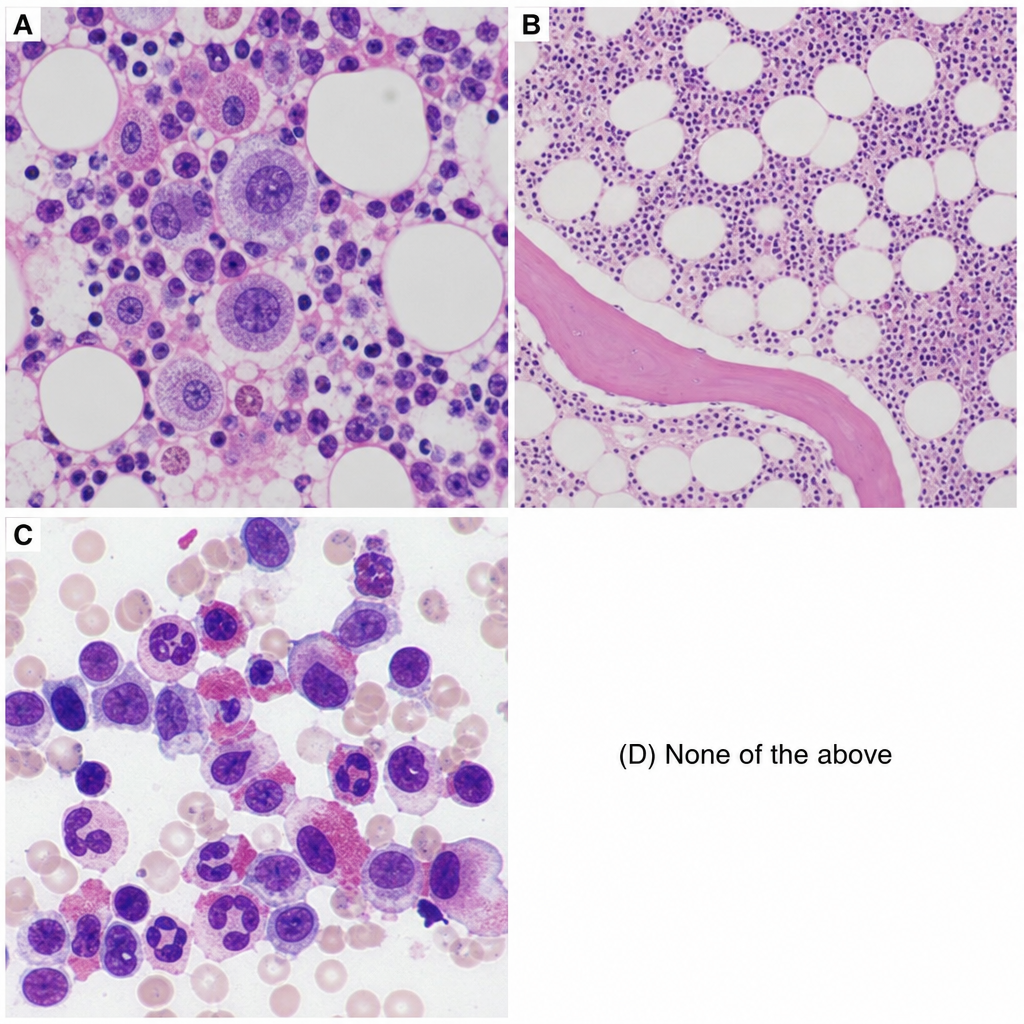
A 56-year-old female presents with headache, dizziness, generalized itching (especially after hot showers), and intense burning in her hands and feet, which is relieved by aspirin. Physical examination reveals splenomegaly and elevated blood pressure. Laboratory findings include HB: 20.1 g/dl, Hematocrit: 60%, WBC: 15,800, Platelet count: 500,000, low EPO, SpO2: 98%, and increased LAP. The patient is positive for JAK2V617F mutation. Which of the following histopathology slides corresponds to this clinical condition?
Bone marrow in Amyloid Lightchain Amyloidosis shows which of the following features?
All of the following blood group systems are known to cause hemolytic disease of the fetus and newborn except?
What is the most common cause of beta thalassemia?
What is the term for localized Langerhans cell histiocytosis affecting the head and neck?
What is the main cause of congestive splenomegaly?
A child presents with Hb of 6.5 gm%, MCV of 65, MCH of 15, and protoporphyria with a much reduced red cell distribution width. What is the most likely diagnosis?
Which of the following is the most important prognostic factor in ALL?
All of the following are true regarding the diagnosis of hemolytic anemia except:
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app