
Hematopathology — MCQs

On this page
The expression of JAK2 mutation is seen in all conditions, EXCEPT:
All of the following are true regarding Chronic Lymphocytic Leukemia (CLL) except?
In which of the following conditions can Downey cells be seen?
All of the following statements about Mucosa Associated Lymphoid Tissue (MALT) lymphomas are true, except:
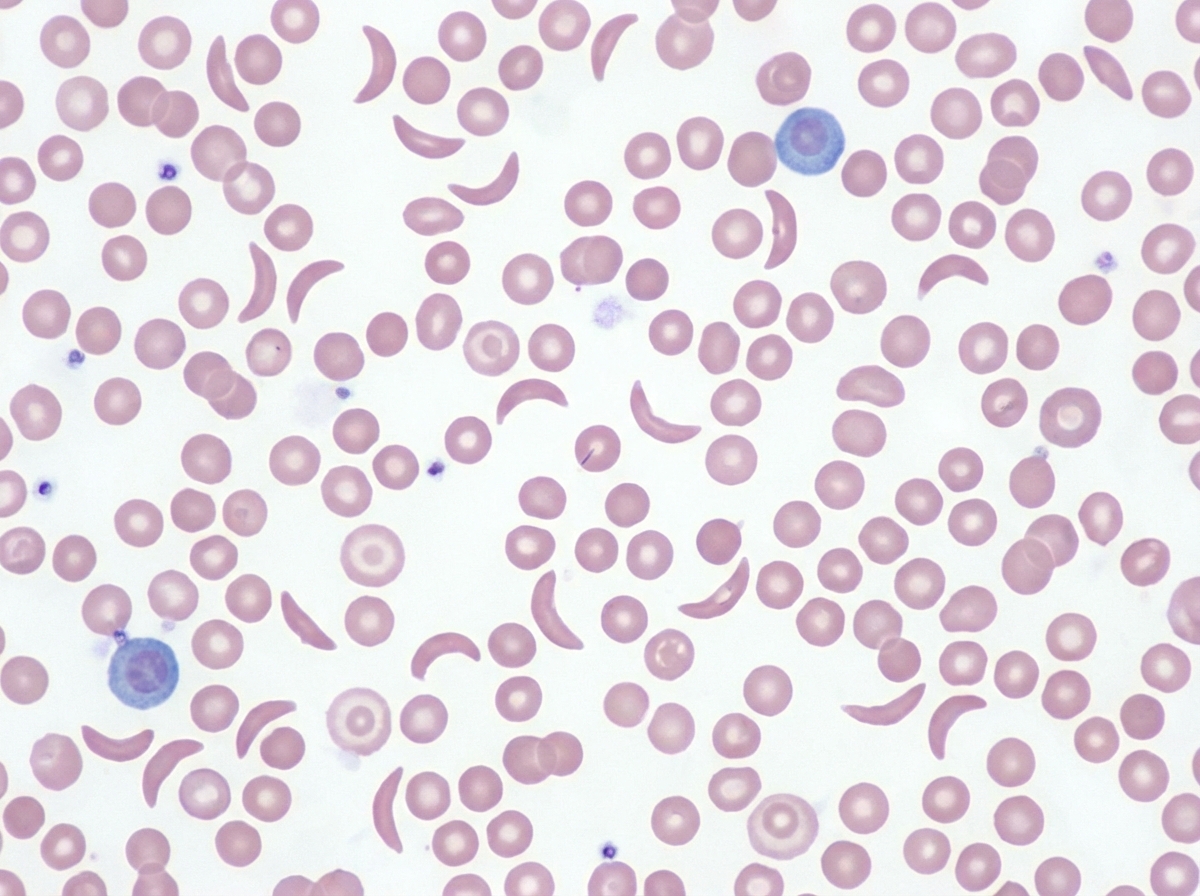
This is the peripheral smear of which disease?
BCR-ABL1 gene fusion is seen in all of the following conditions except?
Which of the following conditions is NOT associated with microcytic hypochromic red blood cells on a peripheral smear?
Which type of Hodgkin's disease is commonest in females and associated with an excellent prognosis?
Which coagulation test is typically prolonged in hemophilia?
Hot agglutinin is found in all conditions except:
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app