
Hematopathology — MCQs

On this page
Which one of the following lymphomas is associated with HTLV-virus infection?
Prognosis of lymphoma depends on all of the following except:
HbH disease is characterized by which of the following genetic defects?
Which of the following features is shared in common between lymphocyte-rich and lymphocyte-predominant types of Hodgkin's lymphoma?
What is true about follicular lymphoma?
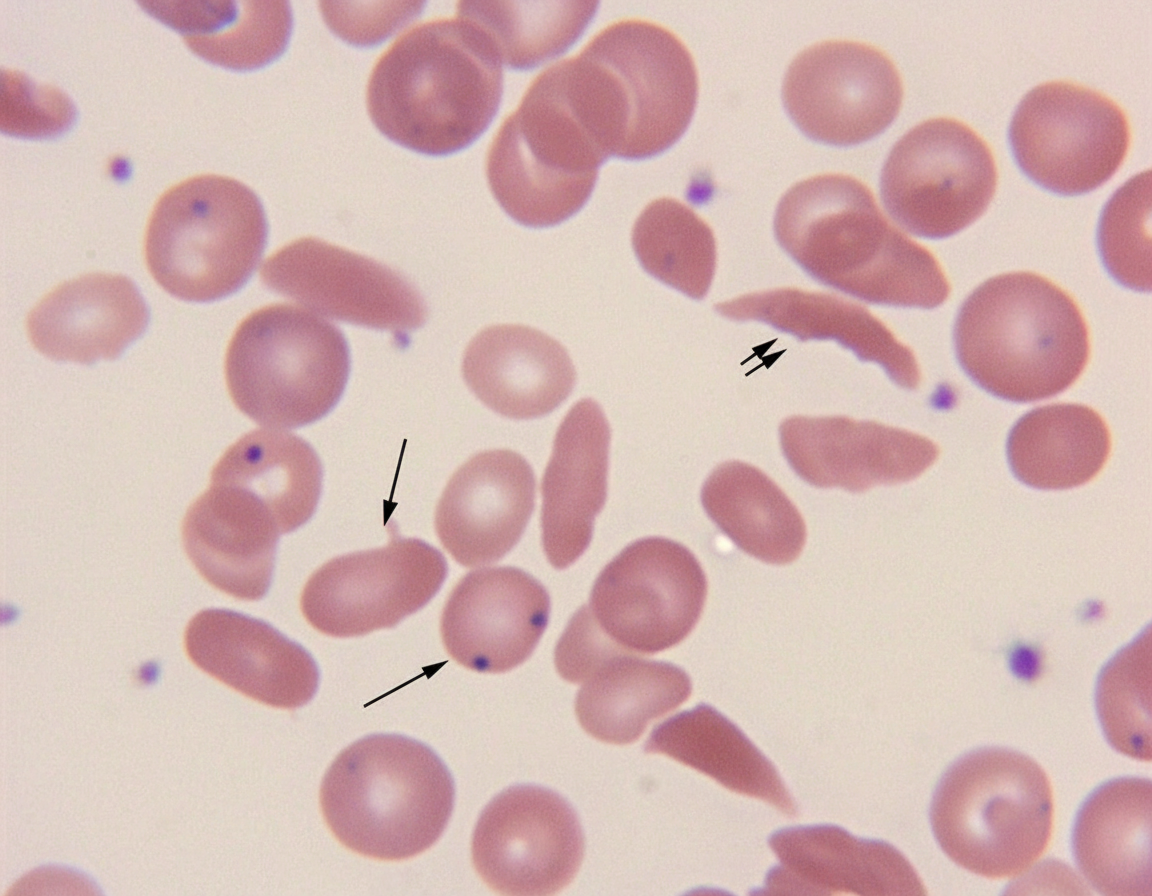
The above marked structures are seen in all except?
Cell of origin of hairy cell leukemia is?
Popcorn cells are characteristic of which of the following hematological malignancies?
Which of the following statements about acute hemolytic reactions is false?
Radiological examination shows evidence of bone infarct in a child. Which of the following conditions may be responsible?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app