
Hematopathology — MCQs

On this page
Testing of recipient cells against donor serum is:
Which of the following tests is used to screen a woman with a family history of thalassemia?
Which one of the following is the most common immunologic type of multiple myeloma?
Which of the following is associated with an intrinsic defect in the RBC membrane?
Massive transfusions result in which of the following complications?
A 48-year-old woman presented with a two-month history of weakness. On examination, cervical lymph nodes were enlarged, and the spleen was palpable 2 cm below the costal margin. The platelet count was 237 x 10^9/L and the total leukocyte count was 40 x 10^9/L, with coarse clumped chromatin noted. Bone marrow revealed a nodular lymphoid infiltrate. Peripheral blood lymphoid cells were negative for CD19, CD5, CD20, and CD23, and also negative for CD79B and FMC-7. What is the most likely diagnosis?
Which of the following is NOT a good prognostic factor for Acute Myeloid Leukemia (AML)?
All of the following statements about Fanconi's anemia are true, EXCEPT?
A 25-year-old female presented with chronic fatigue, hemolytic anemia, cold-induced acrocyanosis, and Raynaud symptoms. Labs show elevated LDH, elevated indirect bilirubin, low haptoglobin, reticulocytosis, and a direct antiglobulin test positive for complement (C3d). A peripheral blood film is shown below. What is the likely cause?
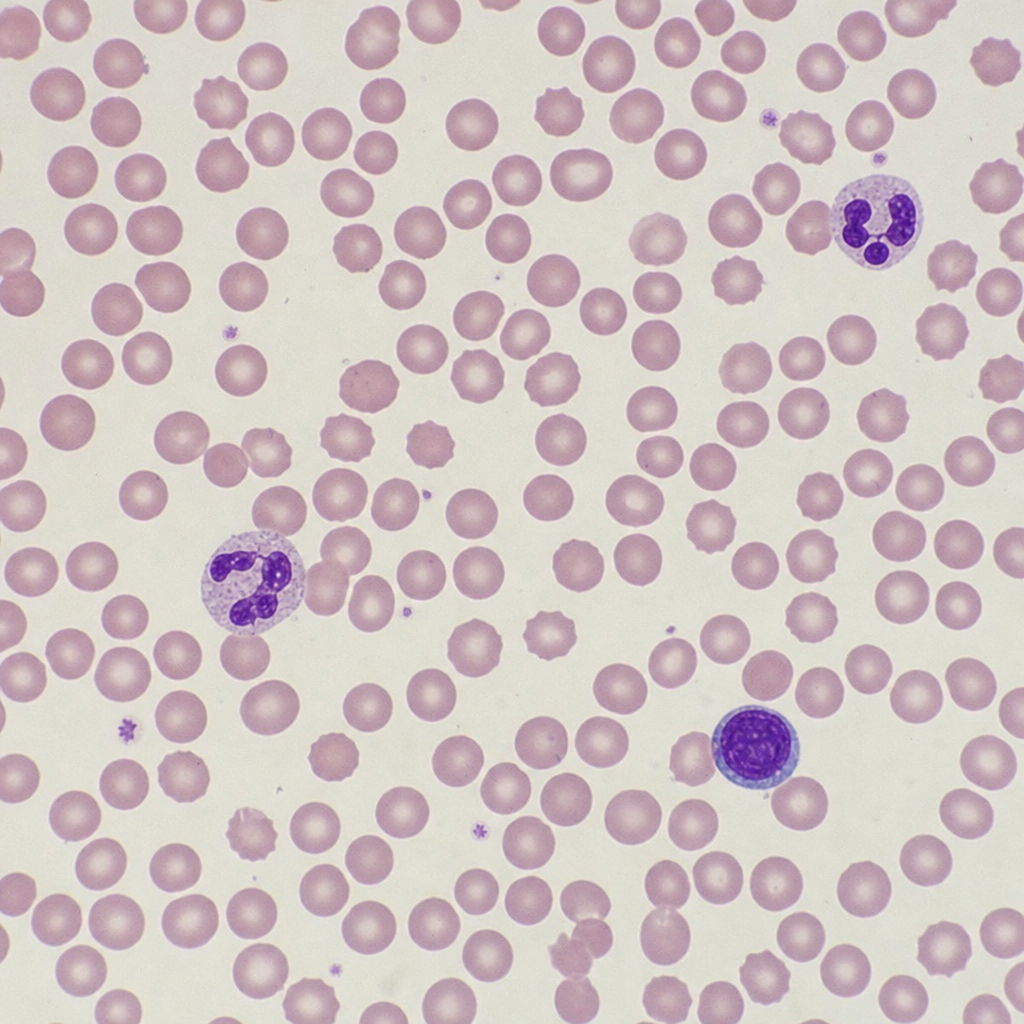
All of the following are true about blood transfusion protocols EXCEPT:
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app