
Hematopathology — MCQs

On this page
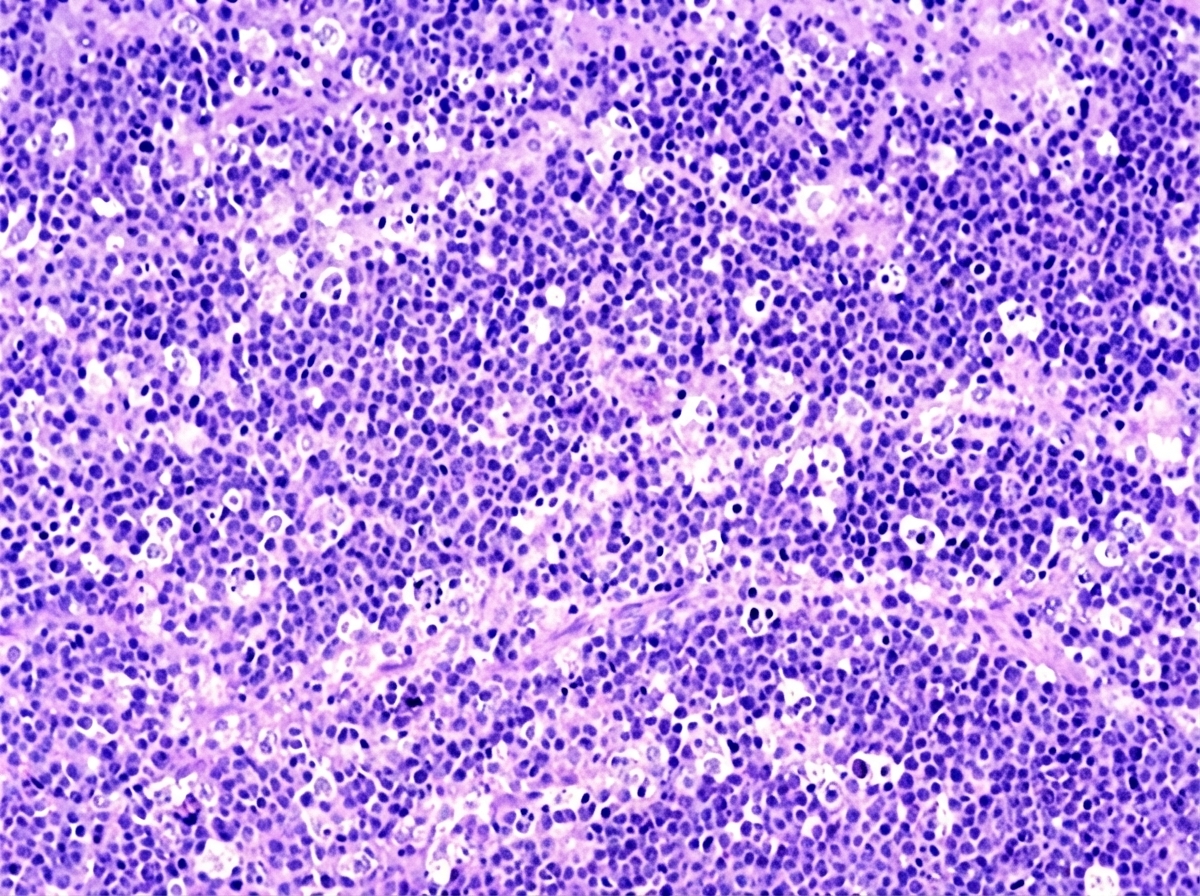
A 4-year-old African boy develops a rapidly enlarging mass that involves the right side of his face. Biopsies of this lesion are shown below. Which chromosomal translocation is associated with this neoplasm?
What is the cause of fragmented RBCs in peripheral blood?
Which of the following is NOT true about Multiple Myeloma?
In hemolytic anemia, what changes occur in the skull bones?
CD19 positive, CD22 positive, CD103 positive monoclonal B-cells with bright kappa positivity were found to comprise 60% of the peripheral blood lymphoid cells on flow cytometric analysis in a 55-year-old man with massive splenomegaly and a total leucocyte count of 3.3 x 10^9/L. Which one of the following is the most likely diagnosis?
Which of the following statements about stored blood is false?
Which of the following is NOT true about Fanconi's anemia?
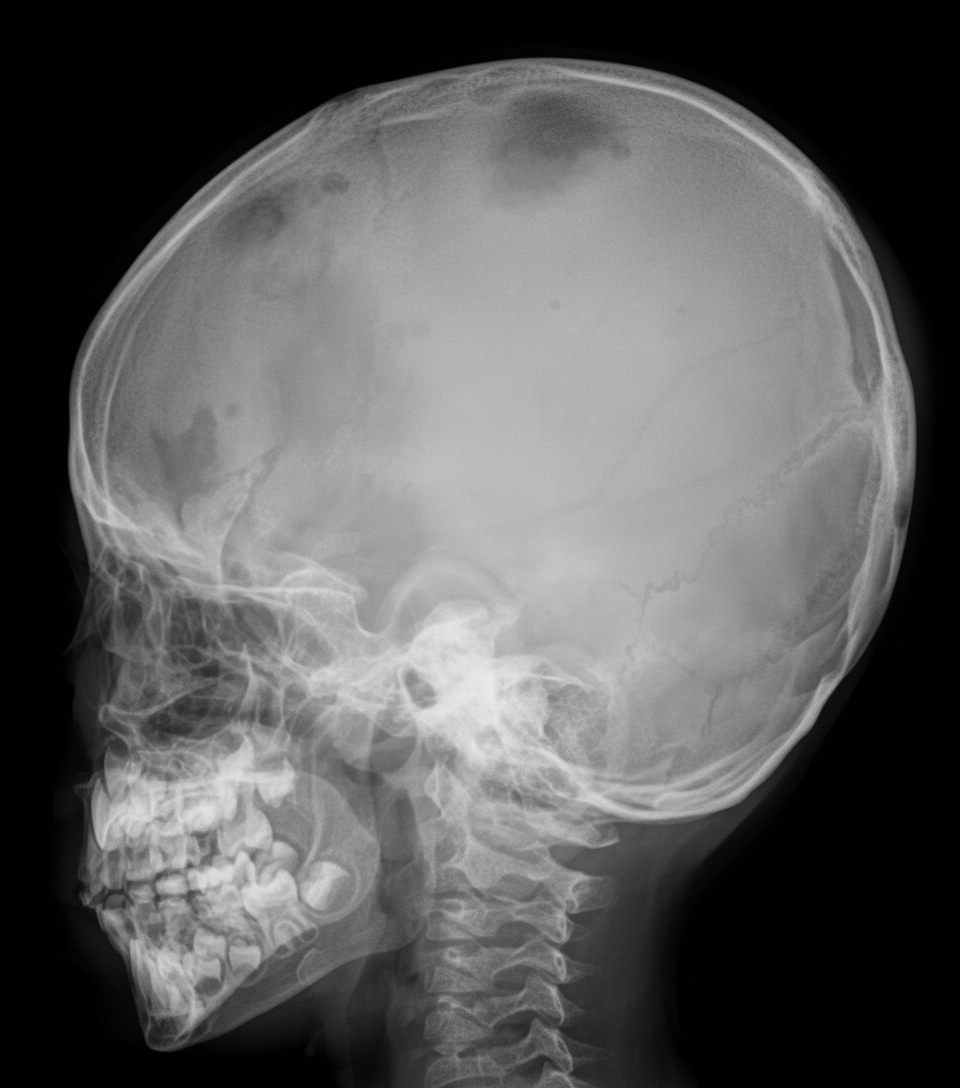
An infant presents with bony pain and dermatitis on the scalp. A skull X-ray is provided. What is the most probable diagnosis?
Which of the following is not a cause of secondary polycythemia?
Which of the following is NOT a cause of Disseminated Intravascular Coagulation (DIC)?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app