
Hematopathology — MCQs

On this page
A 17-year-old woman's urine becomes red after being given sulfonamides for a urinary tract infection. Both urine and serum test positive for free hemoglobin, and the urine red cell count is 1.2 million/mm3. A peripheral blood smear shows normocytic and normochromic red cells and a few "bite cells." Deficiency of which of the following substances is most likely responsible for these symptoms?
A 10-year-old child presents with anemia. Peripheral smear findings are shown below. What is the most probable type of anemia this child is suffering from?
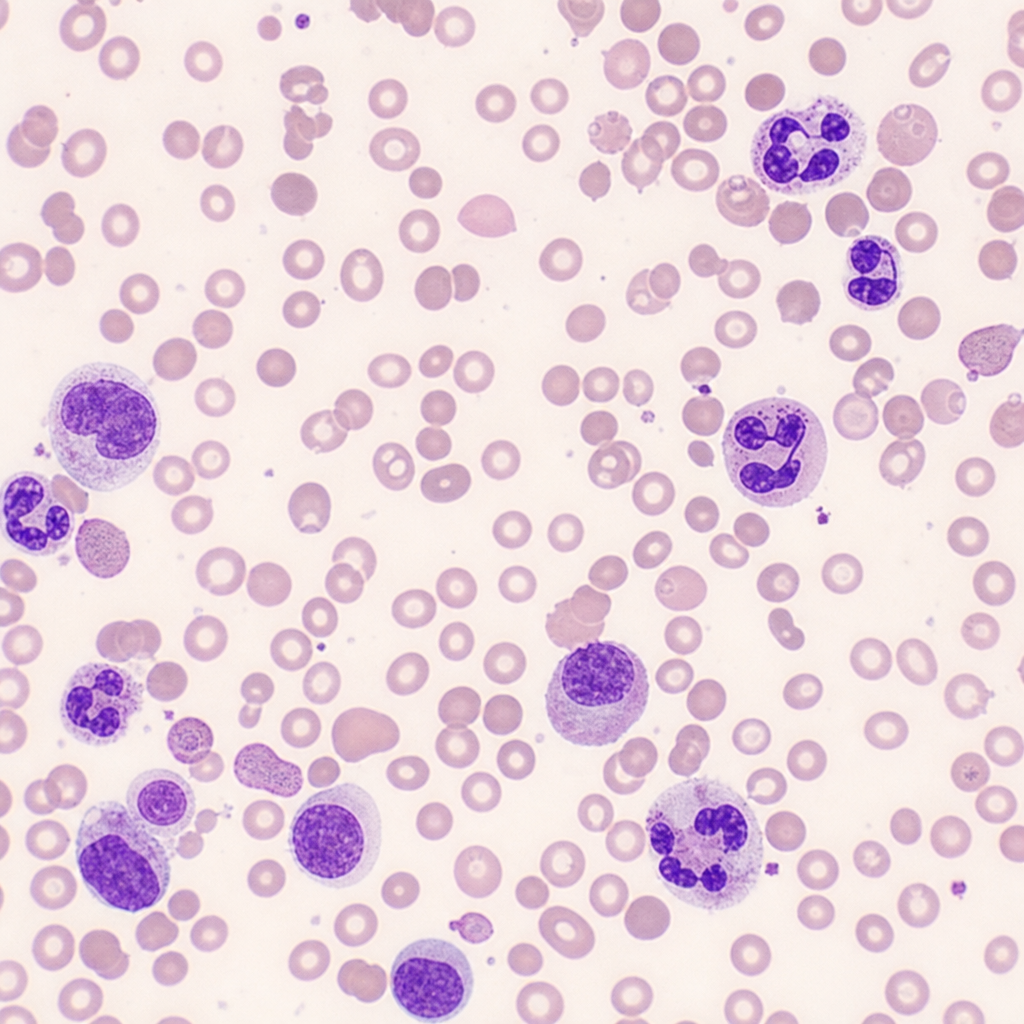
The autohemolysis test is positive in which of the following conditions?
An 8-year-old African boy presents with swelling in his jaw and massive facial disfiguration. Biopsy reveals a tumor invading the bone marrow of the jaw. The pathogenesis of this malignant neoplasm is associated with a virus that exhibits a tropism for which of the following cells?
All of the following are seen in paroxysmal nocturnal hemoglobinuria (PNH) except?
What is the most common subtype of adult acute lymphoblastic leukemia (ALL)?
Beta Thalassemia is best diagnosed by?
Hereditary spherocytosis is characterized by what type of abnormality?
Rate of sickling in sickle cell anemia is directly proportional to all of the following EXCEPT:
Progressive transformation of germinal centers (PTGC) is a precursor lesion of which of the following?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app