
Hematopathology — MCQs

On this page
Features seen in hemolytic anemia are all, EXCEPT:
A patient presents with progressive pallor, hyper-segmented neutrophils, and an MCV > 100 fL. What is the most likely diagnosis?
Which of the following is NOT a B cell marker?
A 4-year-old female is being evaluated for the sudden onset of multiple petechiae and bruises. She is found to have a peripheral leukocyte count of 55,000, 86% of which are small, homogeneous cells that have nuclei with immature chromatin. Indistinct nucleoli are also present. Initial tests on these immature cells are as follows: TdT, positive; PAS, positive; acid phosphatase, positive; and myeloperoxidase, negative. Based on these findings, the immature cells most likely originated from?
Which of the following statements regarding umbilical cord banking is FALSE?
Russel bodies are seen in which of the following conditions?
The type of non-Hodgkin's lymphoma with the highest rate of gastrointestinal system involvement is:
The Coombs test is used for diagnosing which of the following conditions?
Factor IX deficiency results in increased which of the following?
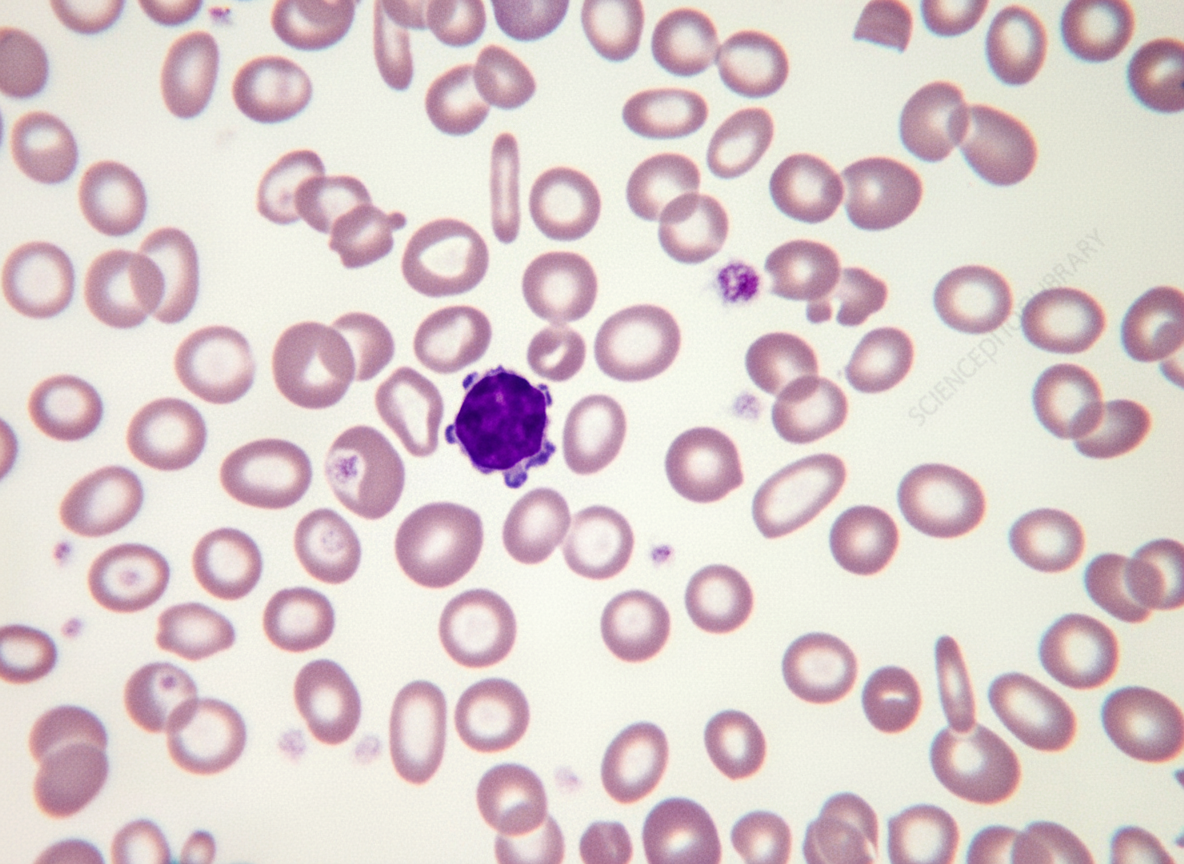
The peripheral blood smear of an anemic 1-year-old child is shown in the illustration. The most likely diagnosis is?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app