
Hematopathology — MCQs

On this page
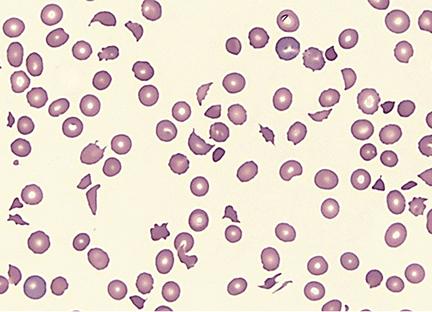
A 2-year-old child in shock presents with multiple non-blanching purple lesions of various sizes scattered on the trunk and extremities. Petechiae are noted, and oozing from the venipuncture site has been observed. The child's peripheral blood smear is shown below. Clotting studies are likely to show which of the following?
Acid Citrate Dextrose (ACD) is used to store blood and preserves Red Blood Cells (RBCs) for 21 days. What are the respective storage periods when phosphate alone is added to ACD, and when adenine and phosphate are added together to ACD?
Which cells are characteristic of Hodgkin's disease?
Non-immune hemolytic anemia occurs in which of the following conditions?
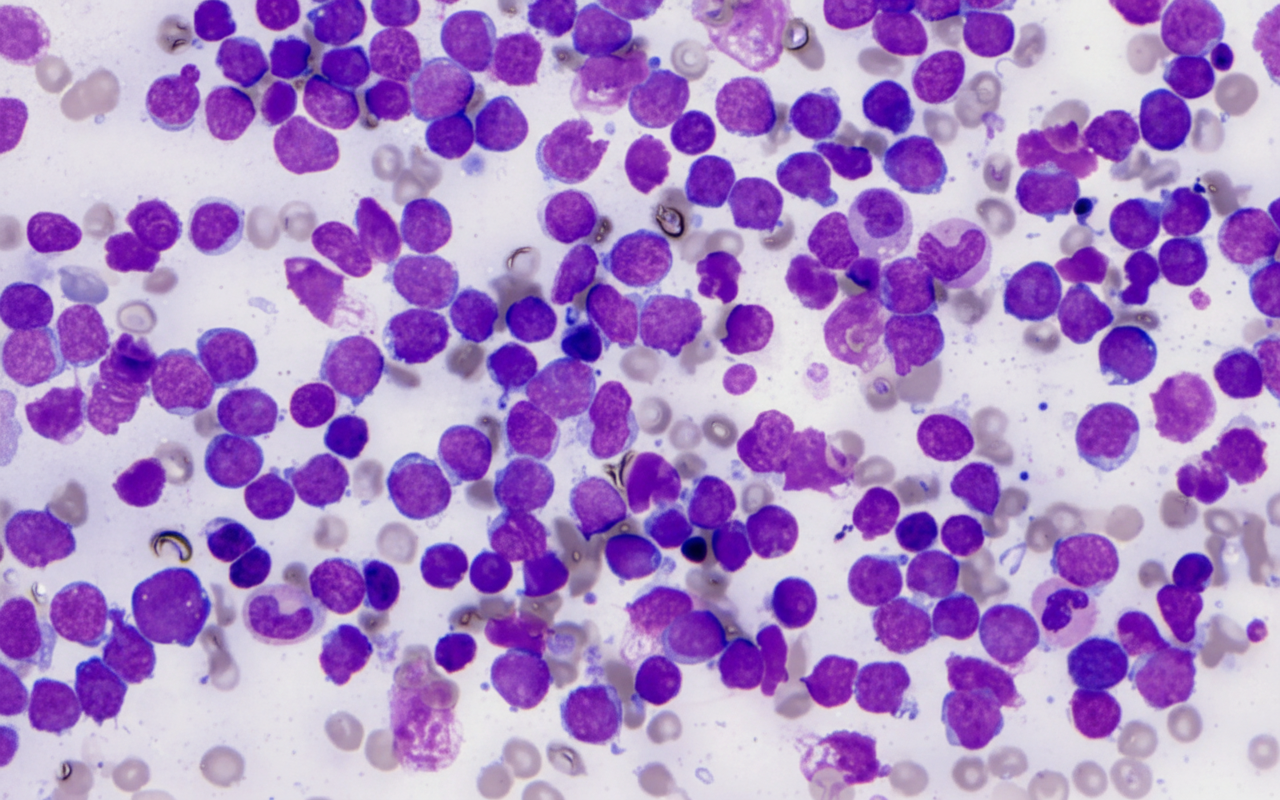
A 7-year-old child presents with fever, bleeding gums, and fatigue for 4 days. On examination, she has splenomegaly. CBC shows Hb of 6 gm%, TLC 75,000/µL, and platelet count of 35,000/µL. Bone marrow aspiration shows these cells comprising 50% of marrow cells. What is the diagnosis?
A patient with Myeloproliferative Neoplasm presents with decreased white cell count and decreased platelets. What is the most likely diagnosis?
Richter transformation is the conversion of?
A thrombotic event is seen in all of the following conditions except?
Which of the following cytogenetic abnormalities is NOT seen in acute myelodysplastic syndrome?
Low TIBC is seen in which of the following conditions?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app