
Hematopathology — MCQs

On this page
Which of the following is absent in cryoprecipitate?
Microangiopathic hemolytic anemia is seen in all of the following diseases except?
Which syndrome is associated with an increased risk of leukemia?
What is the most common cause of aplastic anemia?
TRAP positivity is seen with which of the following hematologic malignancies?
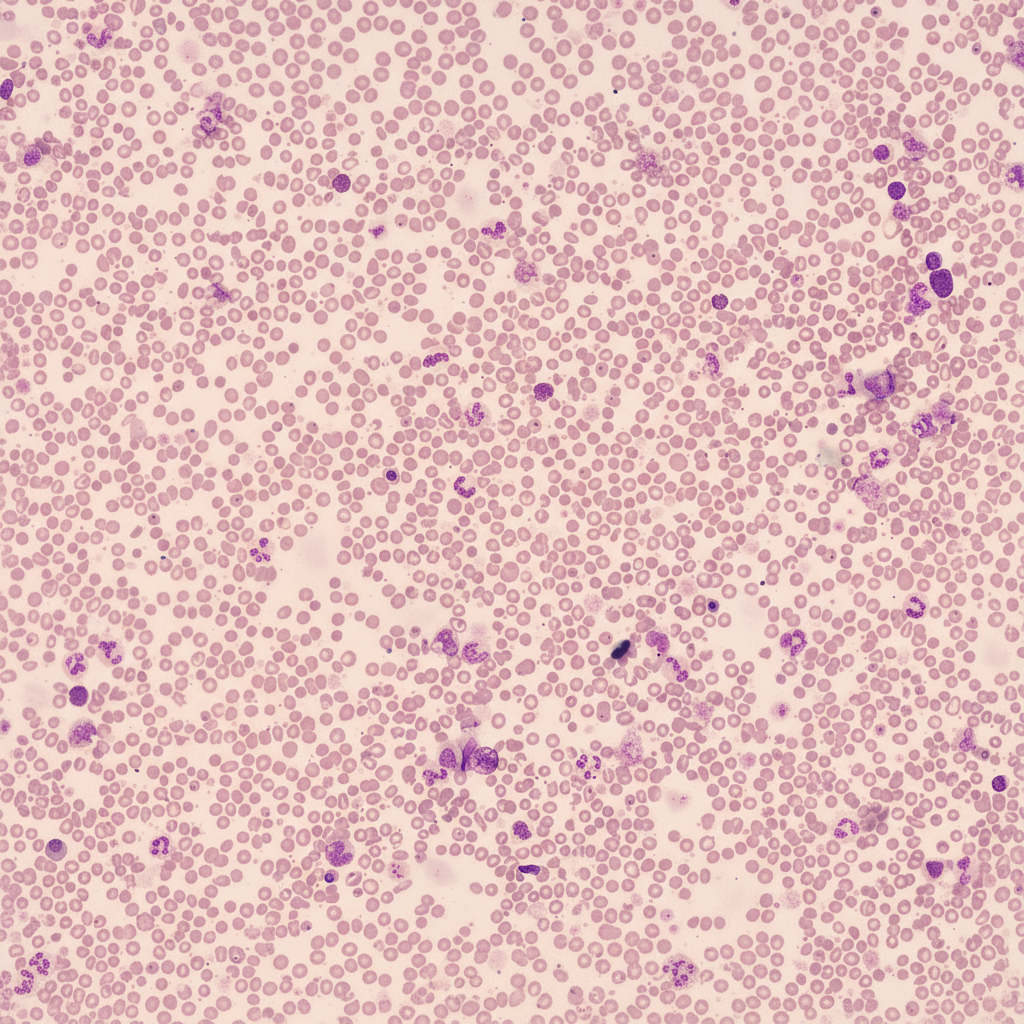
A 30-year-old female with anemia, unresponsive to iron, folic acid, and B12 supplementation, underwent further evaluation. Her CBC profile showed Hb-4.7 g/dL, total WBC count-4,000/mm3, and platelet count-70,000/mm3. Peripheral smear findings are provided. Her bone marrow aspirate revealed a dry tap, and a bone marrow biopsy confirmed the diagnosis. Which of the following stains would have been useful in confirming the bone marrow biopsy findings?
An elderly male presents with headache, recurrent infections, and multiple punched-out lytic lesions on X-ray skull. Which of the following investigations will best help in establishing the diagnosis?
All the following MALT lymphomas are related to a specific organism except:
Pawn ball megakaryocytes are characteristic of which condition?
Which of the following surface glycoproteins is most often expressed in human hematopoietic stem cells?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app