
Hematopathology — MCQs

On this page
Which of the following immunohistochemical stains is used for the diagnosis of lymphomas?
Massive transfusion in a previously healthy adult male can cause hemorrhage due to which of the following mechanisms?
Paroxysmal nocturnal hemoglobinuria (PNH) is associated with a deficiency of which of the following?
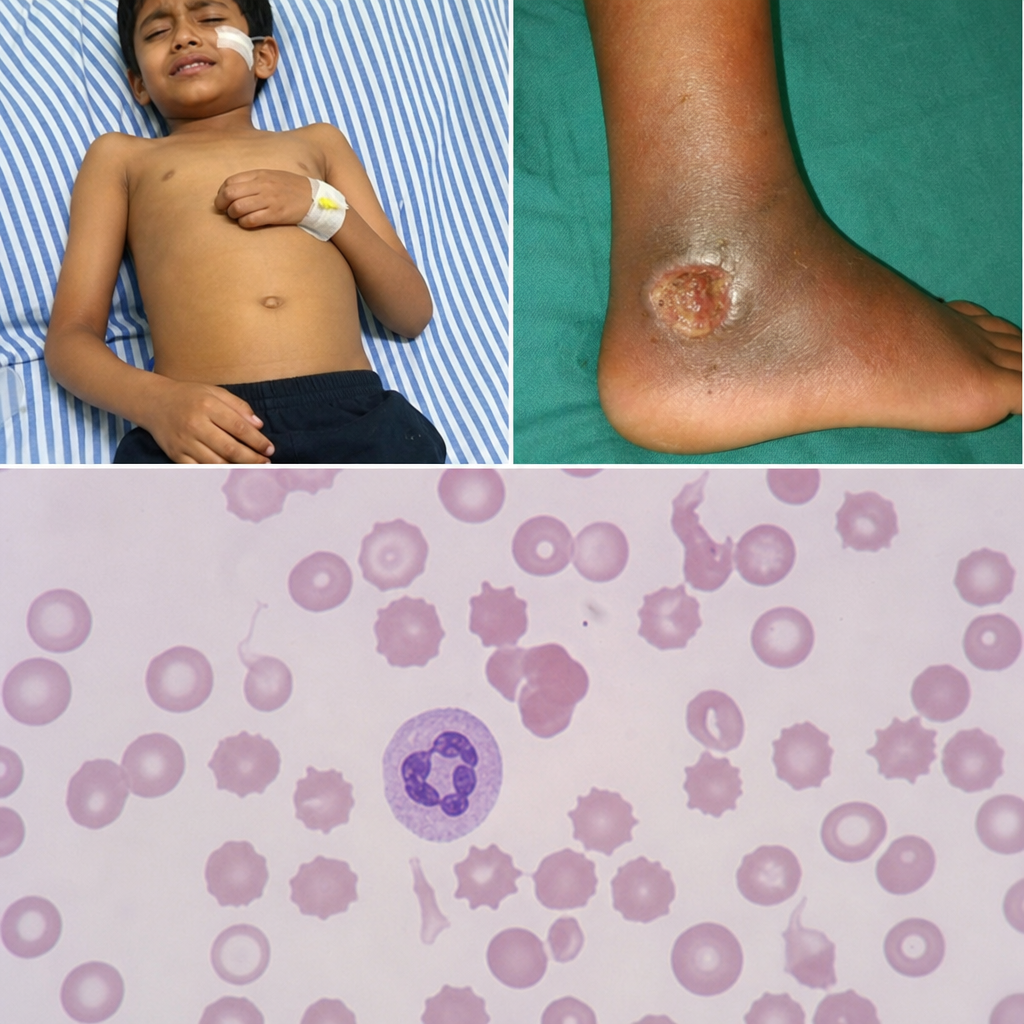
A 6-year-old boy presents with acute abdomen and a nonhealing ulcer on the medial malleolus. A peripheral smear is shown. What is the most likely cause of this condition?
A 24-year-old woman presents with a 4-day history of earache, increased urine production, a skin rash, bone pain on her scalp, otitis media, dermatitis, and exophthalmos. An X-ray of the scalp shows calvarial bone defects, and a fine-needle aspirate displays numerous eosinophils. Which of the following is the most likely diagnosis?
A 6-year-old girl is brought into the emergency room after an automobile accident. Physical examination shows bleeding from multiple wounds, and a CBC reveals a normocytic, normochromic anemia. Which of the following indices is most helpful in defining this patient's anemia as normocytic?
Which of the following may be seen in Multiple Myeloma?
A patient taking an oral sulfonamide develops markedly decreased peripheral blood neutrophil count, while platelets and erythrocytes remain normal. If this neutropenia is caused by antineutrophil antibodies produced in response to the sulfonamide, what would be expected?
A 67-year-old male presents with increasing fatigue and is found to be anemic. Physical examination reveals a hard 1-cm nodule in the left lobe of the prostate. The prostatic-specific antigen (PSA) level is found to be elevated. Examination of the peripheral blood reveals an occasional myelocyte. The erythrocytes are mainly normochromic and normocytic, and teardrop RBCs are not found. There are, however, about two nucleated red blood cells per 100 white cells. What is the best diagnosis for this patient's anemia?
Ring sideroblasts are characteristically seen in which condition?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app