
Hematopathology — MCQs

On this page
What is true about Burkitt's lymphoma?
Which of the following is most likely to cause a hypochromic microcytic anemia?
A 65-year-old man presents with anemia and back pain. A panoramic radiograph reveals multiple radiolucencies. What is the most likely diagnosis?
Which of the following are features of iron deficiency anemia?
Which clotting factor deficiency is asymptomatic?
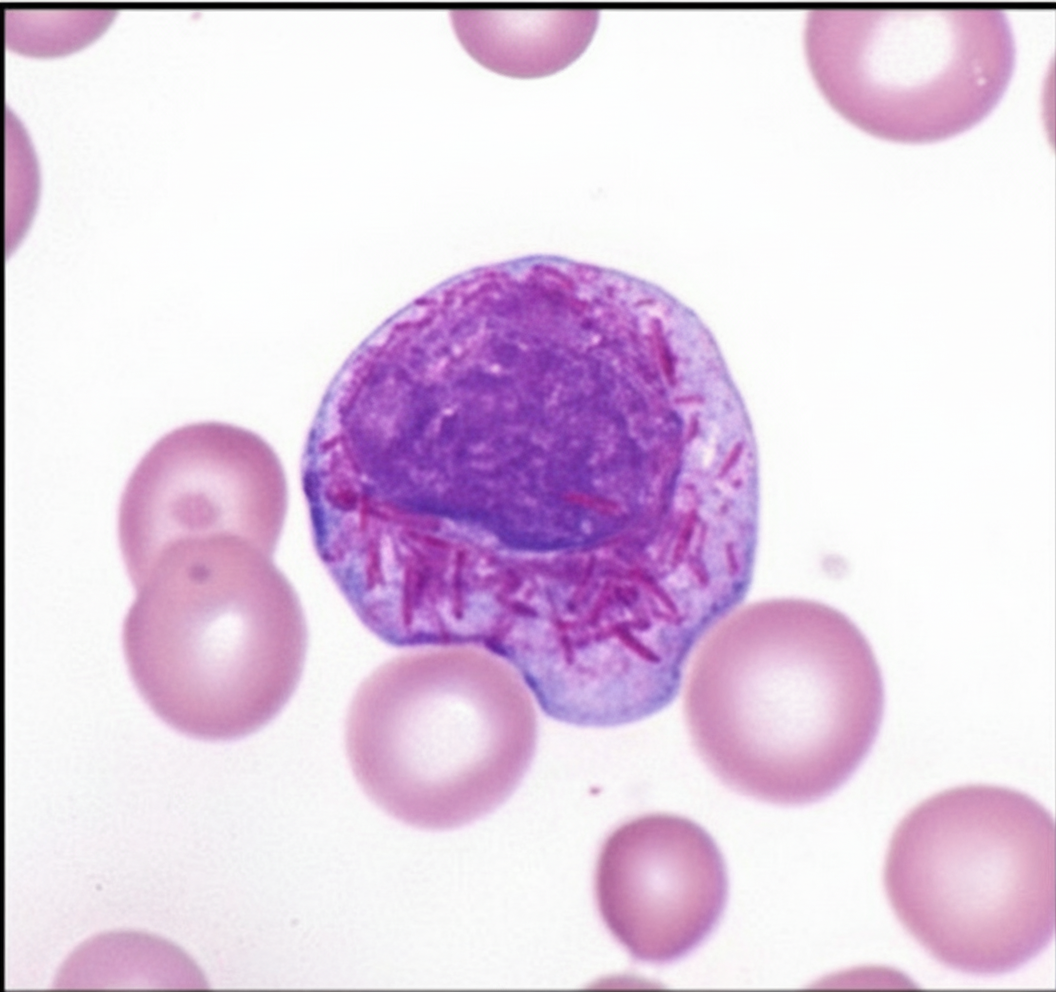
Which of the following hematological malignancies is responsible for the finding shown in the image?
A 9-year-old child presents with continuous bleeding after tonsillectomy. Bleeding time and PTT are prolonged, while platelet count and PT are normal. What is the most likely diagnosis?
A patient of multiple myeloma presents with bony lesions. What is the best prognostic marker for the disease?
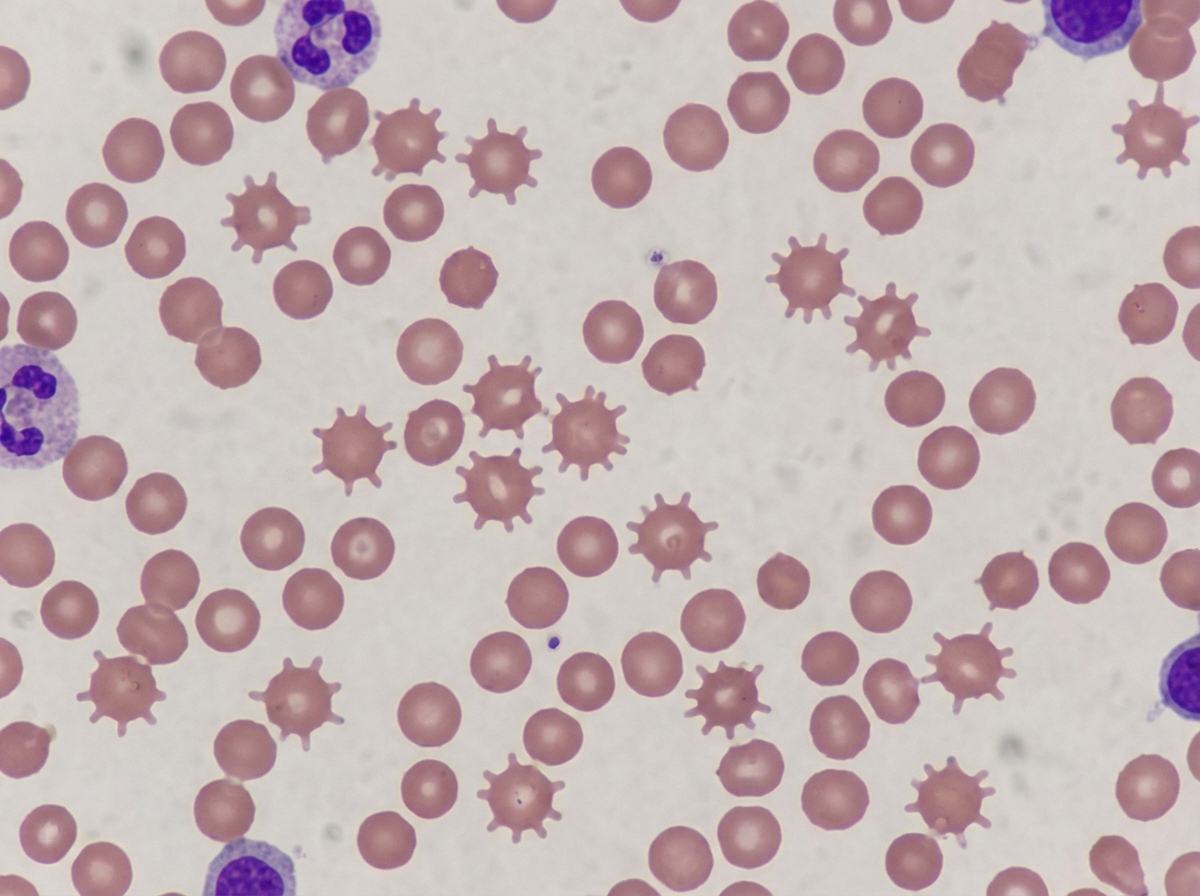
These types of RBCs are seen in which condition?
A 60-year-old male patient complains of bleeding gums and loss of appetite. Histopathologic examination reveals basophilic leukocytosis. What condition might this patient have?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app