
Hematopathology — MCQs

On this page
A patient presents with bleeding due to platelet function defects. Which of the following features is typically observed?
Anemia, splenomegaly, and structural defects are typically seen in which of the following conditions?
Examination of a lymph node from the neck of a 26-year-old man with bilateral cervical lymphadenopathy reveals total effacement of nodal architecture, and at higher power, the characteristic cell shown below. Which of the following additional studies is best suited for confirmation of diagnosis in this case?
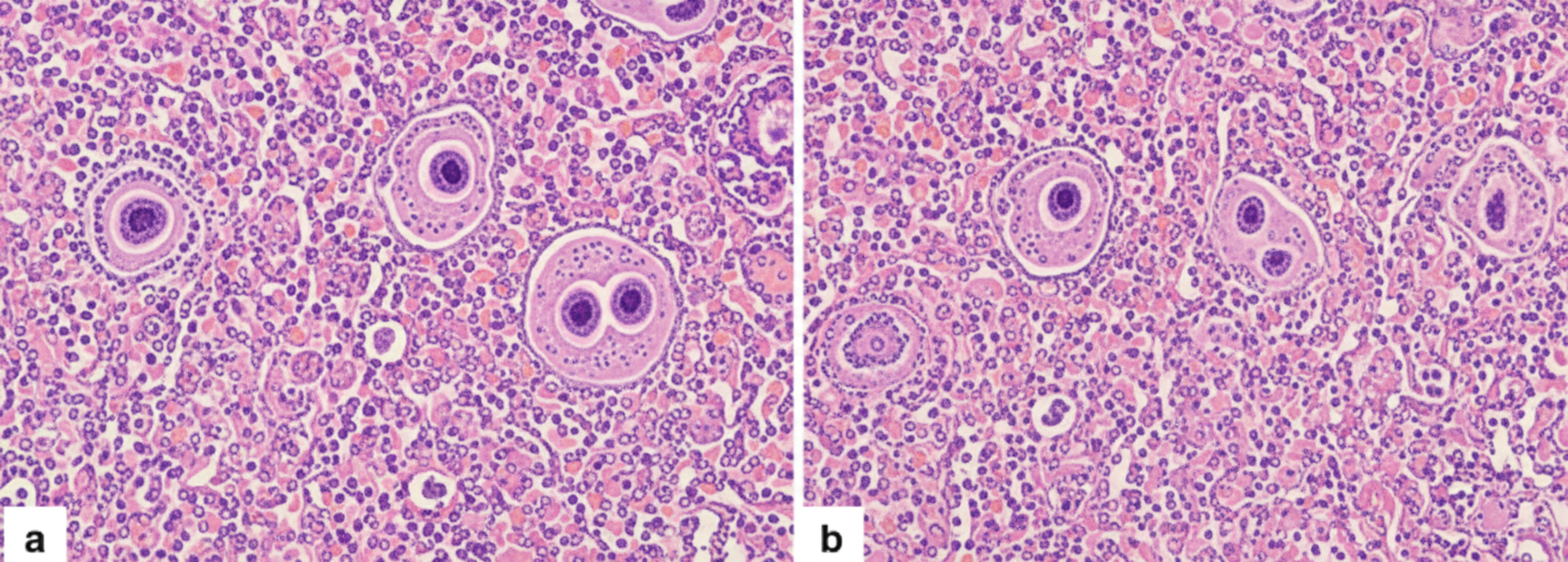
A 28-year-old woman with sickle cell anemia marries a 31-year-old man with no abnormality. What are the respective probabilities of having (i) sickle cell disease and (ii) sickle cell trait in their offspring?
If both parents have sickle cell anemia, what is the likelihood of their offspring inheriting the disease?
A 48-year-old man complains of weakness and easy fatigability for 6 weeks. He has worked for 20 years in a chemical factory that produces a variety of plastics and other synthetic compounds. A complete blood count shows a hemoglobin level of 8.2 g/dL, WBC count of 45,000/mL, and a platelet count of 40,000/mL. Examination of a bone marrow aspirate reveals numerous malignant myeloblasts, and a diagnosis of acute myeloid leukemia is made. Exposure to which of the following agents is the most likely cause of this patient's hematologic disease?
A patient presents with increased aPTT and PT but no bleeding tendency. This was also noted during surgery where no increased bleeding occurred. Which coagulation factor is deficient?
Which gene mutation in AML is associated with a good prognosis, rather than a poor prognosis?
Sezary syndrome is classified under which of the following categories?
A 33-year-old man has experienced multiple nosebleeds along with bleeding gums for the past month. On examination, his temperature is 37.3deg C. He has multiple cutaneous ecchymoses. Laboratory studies show hemoglobin, 8.5 g/dL; hematocrit, 25.7%; platelet count, 13,000/mm3; and WBC count, 52,100/mm3 with 5% segmented neutrophils, 5% bands, 2% myelocytes, 83% blasts, 3% lymphocytes, and 2% monocytes. Examination of his peripheral blood smear shows the blasts have delicate nuclear chromatin along with fine cytoplasmic azurophilic granules. These blasts are CD33+. Which of the following morphologic findings is most likely to be present on his peripheral blood smear?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app